Research Interest
Epigenetic chromatic modifiers in carcinogenesis and inhibitor discovery toward targeted chromatin modifiers
Epigenetic regulation in part involves a dynamic, reversible post-translational modification of histones, which convey inherited information concerning chromatin structure, and dictate the gene expression pattern. Methylation of histone tails has been recently recognized as a key post-translational modification in epigenetics owing to the discovery of histone demethylases. In conjunction with histone methyltransferases (HMTs), these counterpart modifiers work in coordination to maintain a steady global level of methylated histones, mainly at the side chains of lysine (K) and arginine (R) residues of N-terminal unstructured region of histones. Misregulated methylated status of histones from mutational inactivation or abnormal expression of these modifiers has been implicated in oncogenesis.We are recently interested to investigate lysine demethylases of histones (KDMs), focusing on the regulation of post-translational modifications of histones during the initiation and progression of cancer as well as their link to cancer metabolism. We first target a new histone lysine demethylase KDM8 required for cell cycle progression. Structure-based inhibitor discovery toward KDMs is also underway.
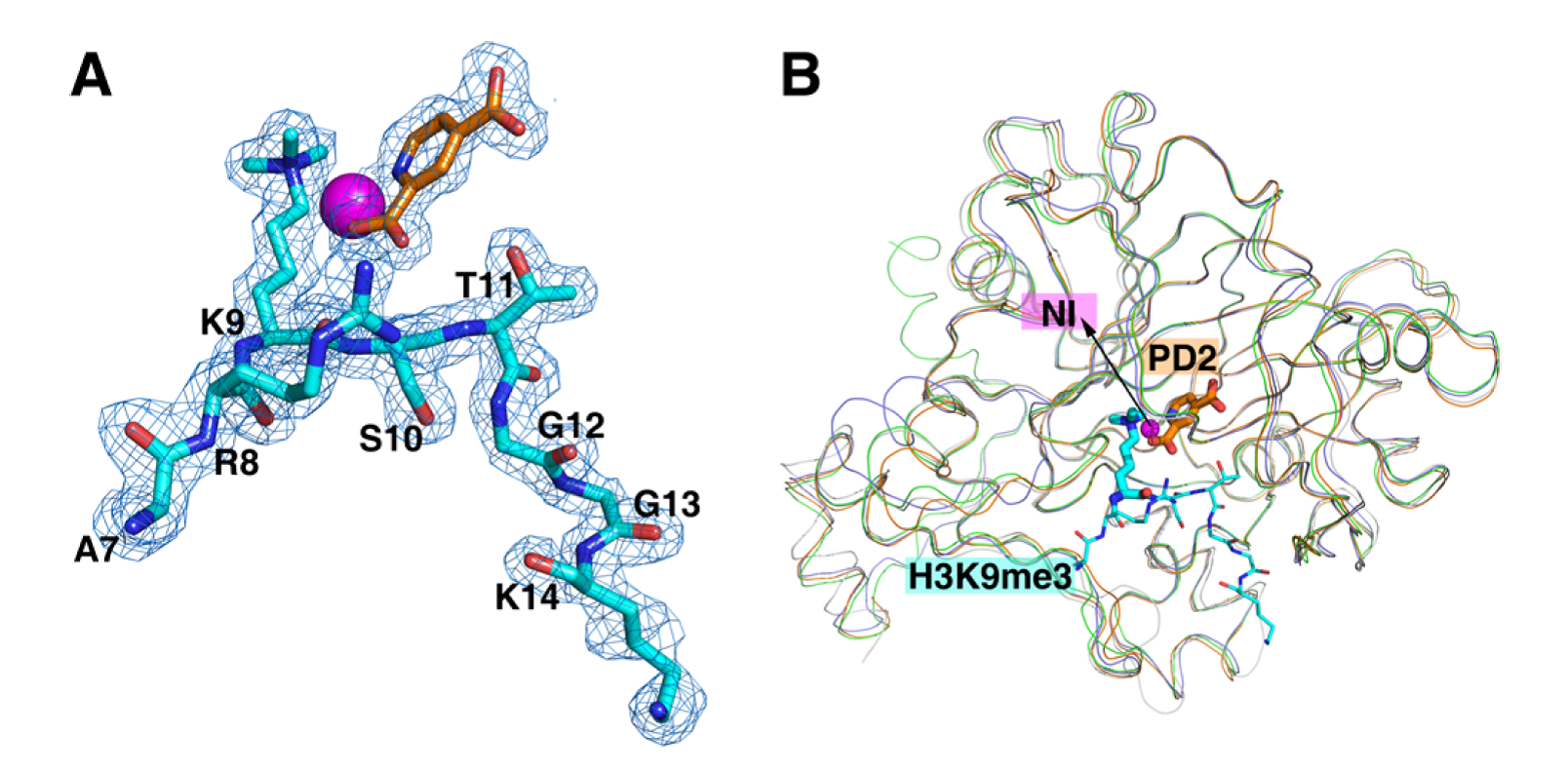
Figure 1. Crystal structure of the catalytic domain of histone lysine demethylase KDM4B. (A) The 2Fo-Fc electron density map of inhibitor PD2 (orange), Ni2+ ion (magenta), and the substrate peptide (cyan), where lysine 9 (K9) is trimethylated. (B) Structure comparison of KDM4A (PDB code: 2OQ6), KDM4B (this study; PDB code: 4LXL), KDM4C (PDB code: 2XML), and KDM4D (PDB code: 4HON). Adapted from Chu et al., J. Med. Chem., 2014.

Regulation of key metabolic enzymes lead to alteration of cellular energy flux and gene expression pattern in cancer cells
One
of the hallmarks of cancer cells is their altered metabolism to sustain their
high proliferation rate referred to as aerobic glycolysis, or the Warburg
effect. The
tumor-preferred pathway involves an increased uptake of glucose, utilization of
intracellular glucose to pyruvate via glycolysis, and the conversion into
lactate in the presence of sufficient oxygen. Along this metabolic flux, PKM2
that catalyzes the dephosphorylation of phosphoenolpyruvate (PEP) to pyruvate,
is a pivotal enzyme and selectively expressed in tumors cells. Its abundance
and activity determine the metabolic flow to lactate, TCA cycle or biosynthetic
pathway. There are four isoforms of pyruvate kinase in
mammals: tissue-specific PKL and PKR encoded by PKLR, and PKM1 and PKM2
that are mutually exclusive products of PKM. Of note, PKM2 is expressed
in highly multiplying cells including embryonic, adult stem cells, and
re-expressed in tumor cells, while PKM1 expression is predominantly found in
heart, brain and muscle cells that demand high ATP, suggesting that cancer cells favor the
expression of PKM2
There
are two ways that PKM2 pyruvate kinase activity can be modulated: 1) by
metabolites such as FBP, serine and SAICAR as activators while Phe, Cys and T3
as negative regulators; and 2) by post-translational modifications: phosphorylation of Tyr105 by growth factor
signals, oxidation of Cys358 by ROS, acetylation of Lys305 and Lys433 by high
glucose. In most
of these cases, suppression of PKM2 pyruvate kinase activities are favored in
response to oncogenic signals.
Yet, another remarkable way of suppressing
cytosolic PKM2 activity is its translocation into nucleus, where
it serves as transcriptional coactivator or as a protein kinase to
modulate transcriptional program. Nuclear translocation of PKM2 would stop the
glycolysis flow at PEP and accumulate intermediates, which are often precursors
for biosynthesis to increase biomass. Further, nuclear PKM2’s protein kinase
activities are found to phosphorylate nuclear proteins H3 and Stat3. As a
transcriptional activator, it induces metabolic and oncogenic genes.
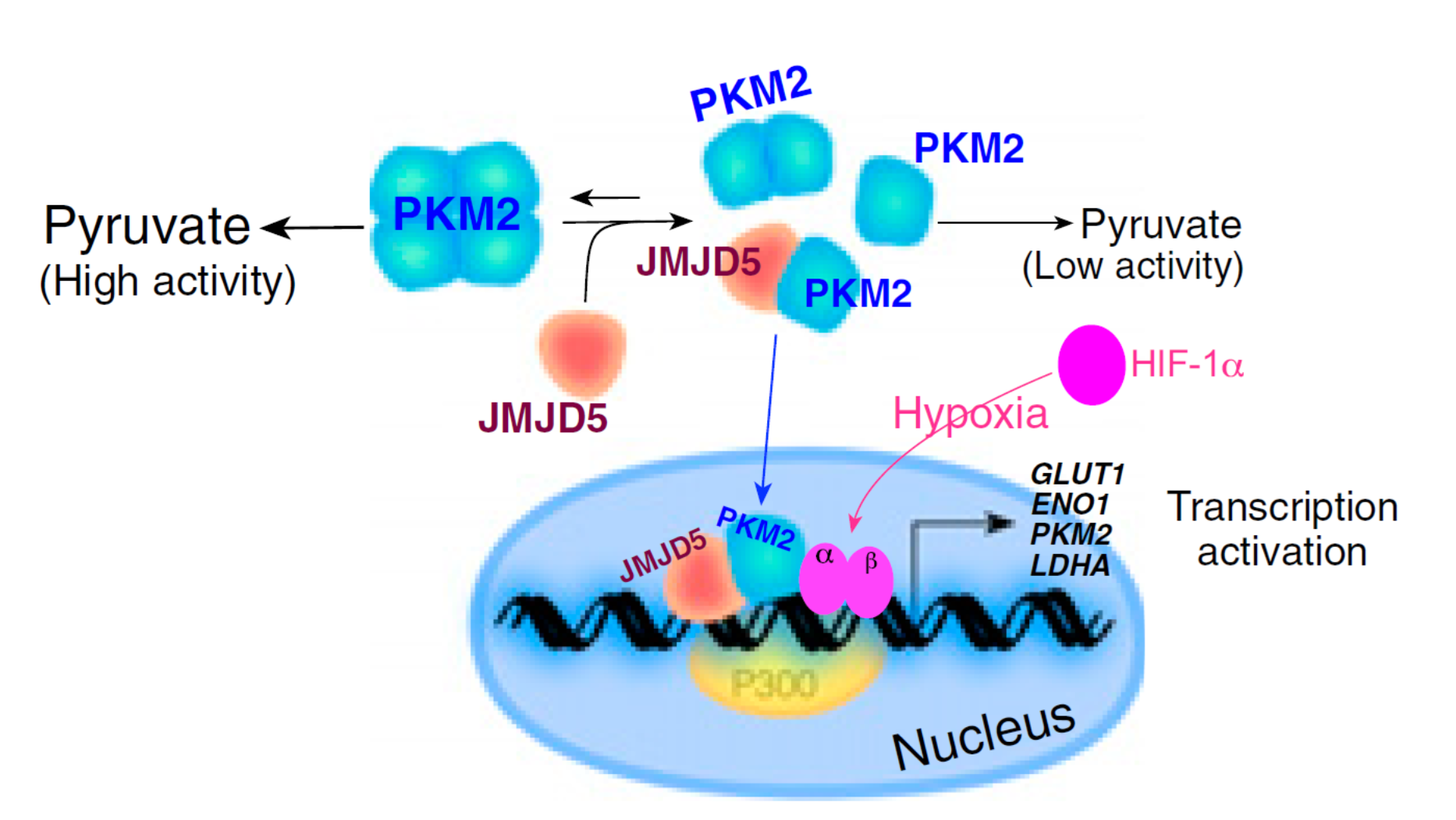
Figure 2. The proposed model that depicts JMJD5 as a major regulator in PKM2-stimulated HIF-1α metabolic reprogramming. JMJD5 and PKM2 are corecruited to HREs of LDHA and thereby specifically enhance HIF-1α binding.
The PI’s lab has been working in the molecular pathogenesis of Helicobacter pylori that leads to severe gastrointestinal diseases including gastric cancers. We have focused on several virulence factors including VacA, CagA, the blood group antigen-binding adhesion (BabA2) and more recently, cholesterol-α-glucosyltransferase involved in adhesion, invasion, as well as underlying mechanisms in hijacking host-cell signaling. We have also reported antimicrobial resistance in H. pylori isolates in Taiwan, the relationship of IL-1b and IL-10 polymorphisms and H. pylori infection with erosive reflux esophagitis and gastritis and the expression of Foxp3+ Treg that was positively associated with the severity of gastroduodenal diseases.
Target-based Drug Discovery toward the Shikimate
Pathway Enzymes of Helicobacter pylori
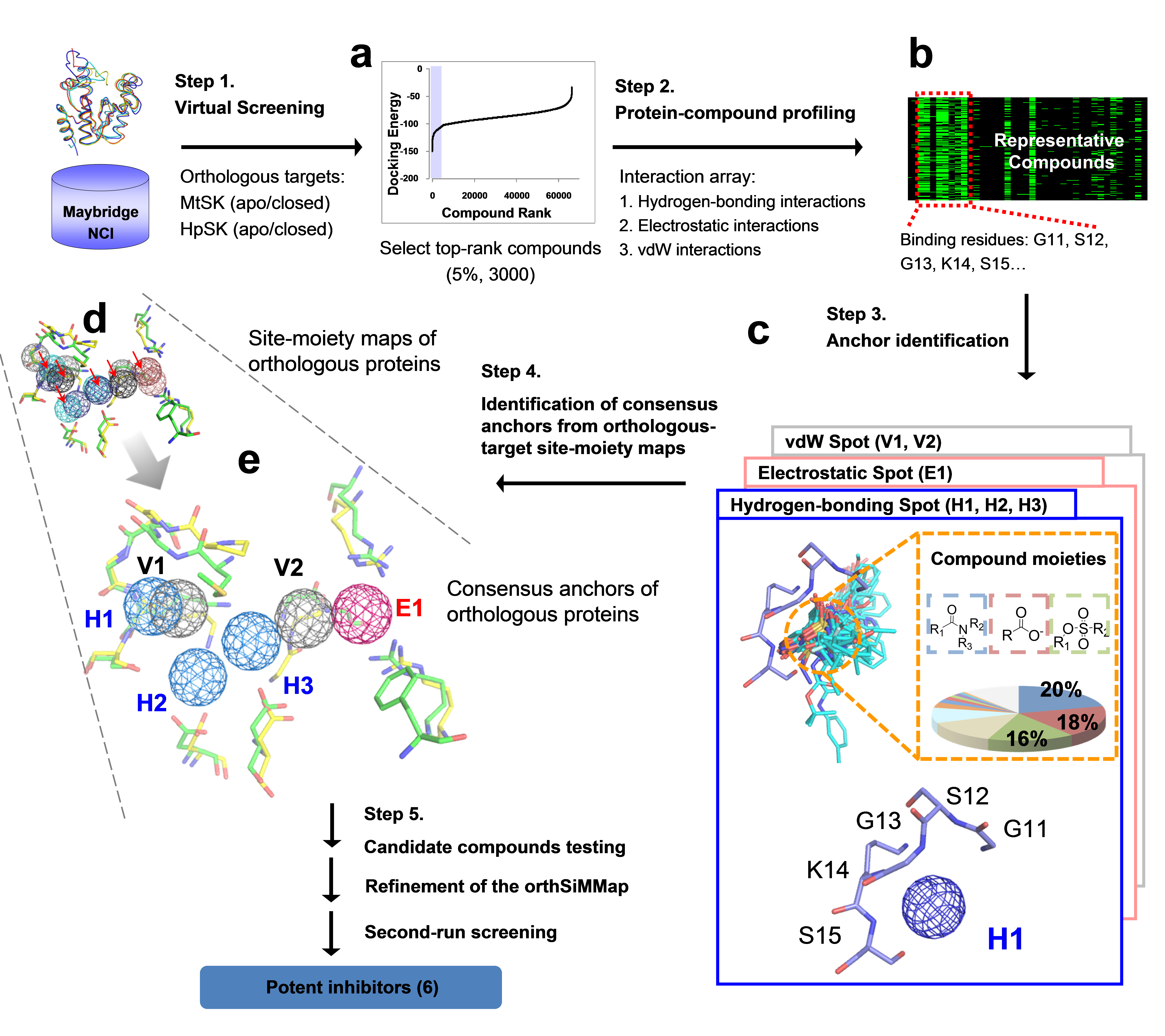
Figure 4. Framework of the orthSiMMap-based screening method. In Step 1, GEMDOCK was used to generate docked poses for HpSK and MtSK by screening compound libraries (Maybridge and NCI). For each target (HpSK or MtSK), the protein-compound interacting profile was derived from fusing the top ranked 2% (~6,000) compounds. In Step 3, conserved interactions of the target protein and chemical moieties of ligands are identified to deduce the anchors of HpSK and MtSK. The orthSiMMap is constructed based on the conserved features between orthologous target site-moiety maps, which will be used to select candidate compounds for the enzymatic assay. Finally, the model is refined based on the bioassay of candidate compounds.