- Mitochondrial complex I and diseases
My research interests are centered on understanding the mechanisms of complex I and developing a strategy to reestablish the functionality of complex I thus lead to the treatment of human mitochondrial diseases caused by the defects of this enzyme complex.
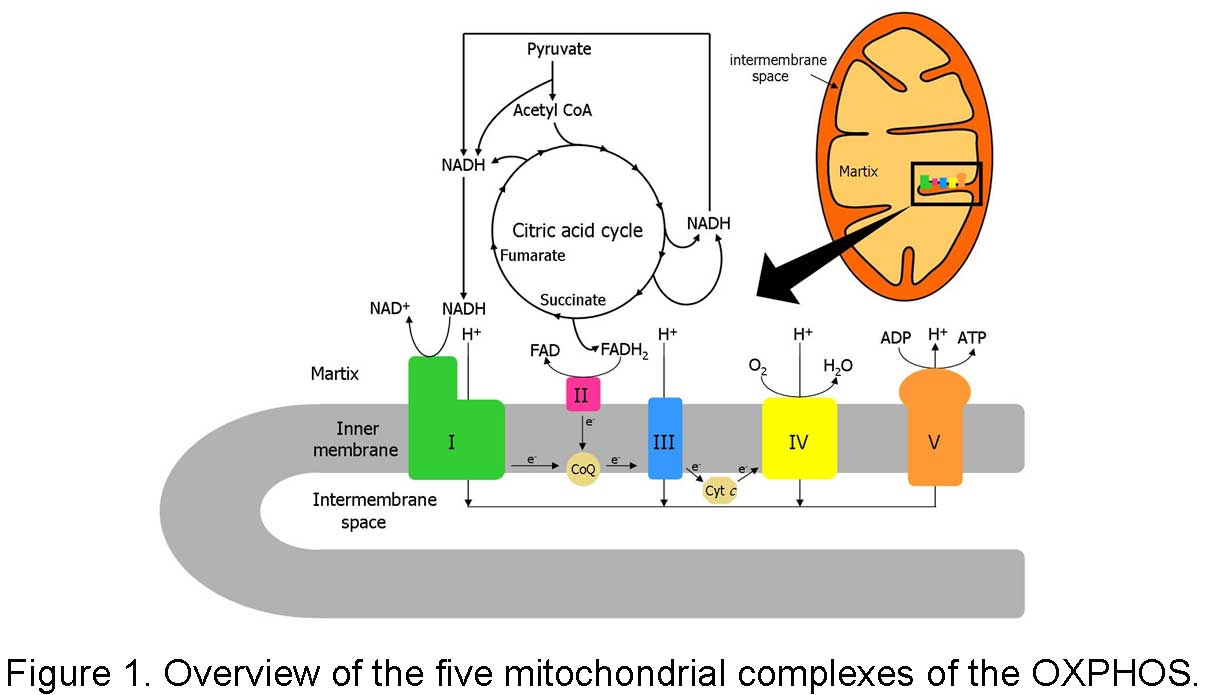
Human complex I is the largest, most intricate and least understood respiratory complex in mitochondrial oxidative phosphorylation (OXPHOS) system, and is composed of at least 45 unlike subunits. Of these, 7 subunits, including ND1-6 and ND4L, are encoded by mitochondrial DNA (mtDNA), and the other 38 subunits are encoded by nuclear DNA (nDNA). It is the point of entry for the major fraction of electrons that traverse the respiratory chain eventually resulting in the reduction of oxygen. Coupled to the electron transfer, protons are pumped from the matrix side to the intermembrane space of mitochondria. In contrast, the bacterial version of complex I, also called NDH-1, is located in the plasma membrane and contains only 13 or 14 different subunits. Despite the difference in the size and complexity, complex I and NDH-1 share many properties in terms of structure and function.
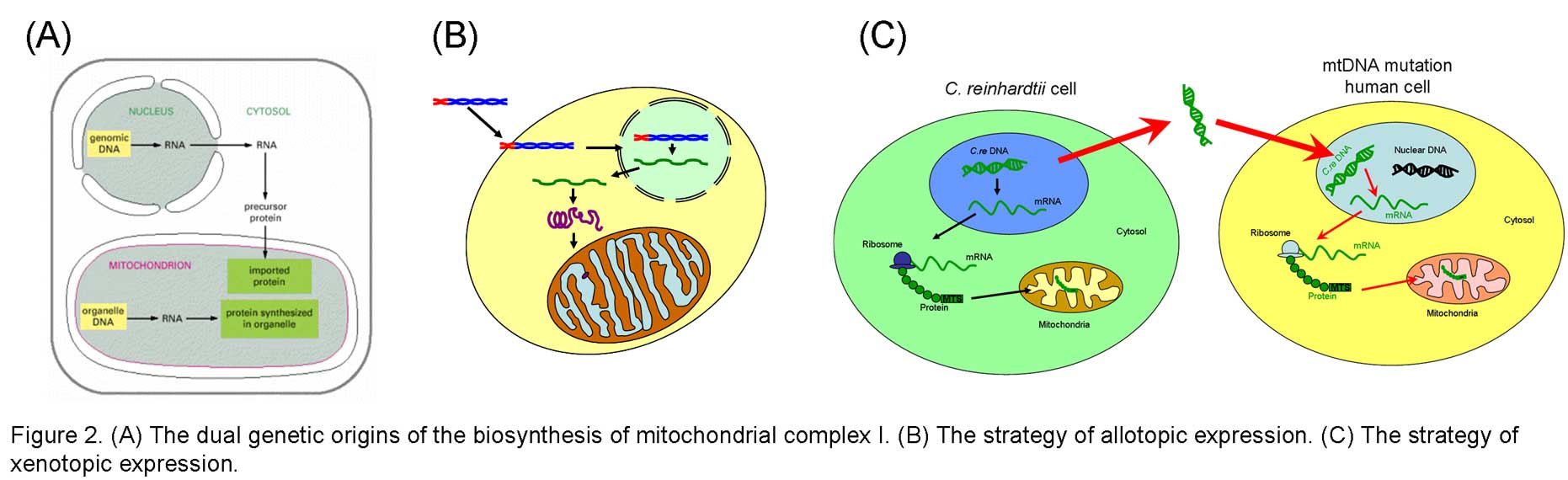
In eukaryotes, it is not easy to manipulate mitochondrial DNA directly. In contrast, gene manipulation procedures of bacterial DNA are well advanced. Previously, we have used Paracoccus denitrificans and Escherichia coli NDH-1 as models to study the function-structure relations of this complex and investigated the events that are associated with pathogenic mutations of human complex I. Our discoveries from these studies demonstrate that bacterial model systems could be useful for further understanding of this complicated mammalian enzyme, and provide an alternative way to gain insight into human pathogenesis derived from malfunction of this complex. However, not every aspect of human diseases can be reflected in the bacterial system. Therefore, a better understanding of complex I in mammalian mitochondria is absolutely necessary.
Dysfunction of complex I has been linked to a wide variety of mitochondrial diseases, including Leber Hereditary Optic Neuropathy (LHON) and Parkinson’s disease, as well as the process of aging. Because of the complexity of the enzyme complex and its dual genetic origin, the pathogenic mechanism of most complex I defects is still unknown, and no effective remedies have been established for complex I deficiencies. From this point of view, developing methods to correct complex I defects seems important. The specific aims of our studies are as the followings:
- Understanding the functional mechanisms of human complex I.
- Revealing the functional and structural roles of component subunits in complex I and their association with mitochondrial diseases.
- Developing new approaches to treat the diseases caused by the defects of complex I.
- Allotopic expression of mitochondrial-encoded subunits of complex I
- Xenotopic expression of mitochondrial-encoded subunits of complex I
- The strategy of organelle-targeted delivery of mitochondrial proteins using the protein transduction domain (PTD)
- Gene therapy of nuclear- encoded subunits of complex I
2. Helicobacter pylori pathogenesis and persistence
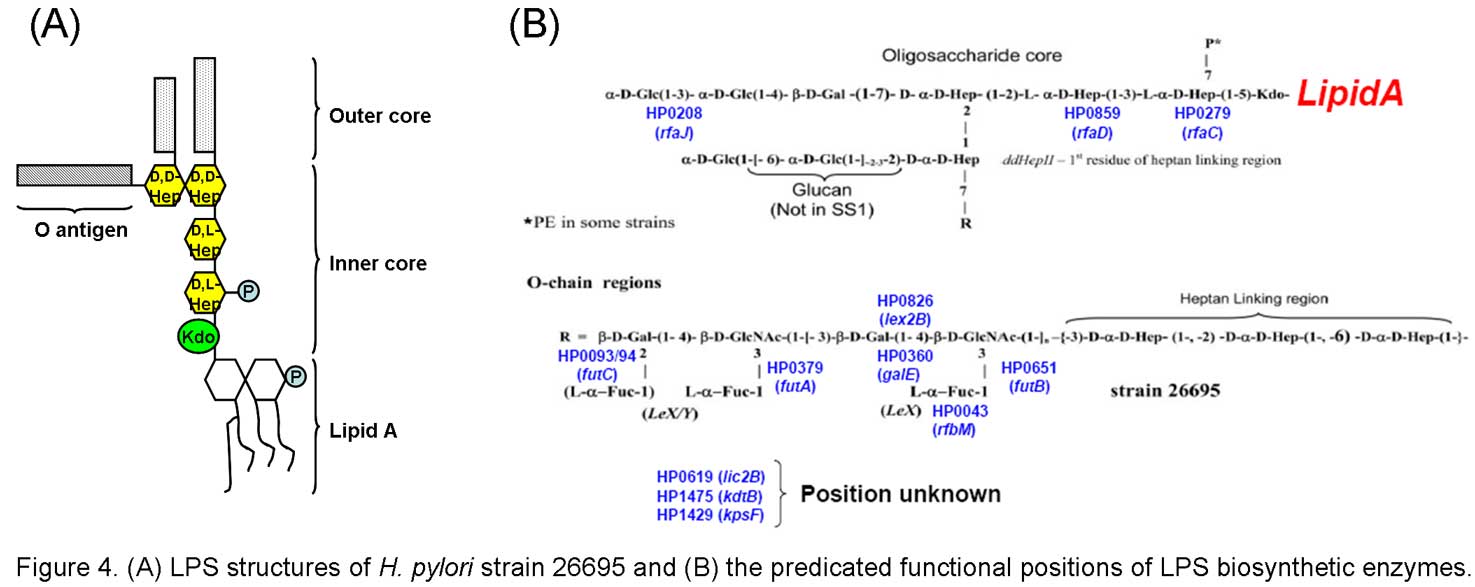
Helicobacter pylori infection affects more than 50% of world’s population and is associated with a wide spectrum of clinical outcomes including gastritis, peptic ulcer and gastric cancer. In spite of the identification of numerous virulence factors, the precise mechanism of pathogenesis of this bacterium remains poorly understood. Lipopolysaccharide (LPS) is a major surface antigen of gram-negative bacteria and is essential for the physical integrity and function of the bacterial outer membrane. In addition to contributing to host evasion or autoimmunity, recent data have suggested that the LPS in H. pylori may actually mediate adhesion and is responsible for phase variation. Although considerable effort has been invested in identifying the genetic determinants of LPS biosynthesis, some key biosynthetic enzymes have yet to be identified. Therefore, to have a better understanding the role played by LPS in the virulence of H. pylori, studying on the enzymes involved in the biosynthesis of H. pylori LPS seems important.
In our laboratory, we aim to apply genetic, biochemical, bioinformatic, immunological and structural approaches to elucidate the genetic basis of LPS biosynthesis in H. pylori.Several open reading frames (ORFs) which have been identified by homology with other polysaccharide biosynthesis genes and have been implicated in H. pylori LPS synthesis are chosen as the primary targets. The respective gene products will be structurally determined and functionally characterized. In addition, the purified LPSs from mutants generated by insertion inactivation of the targeting genes will be analyzed in detail, and their influence on host cell infection will also be studied at cell culture level. Furthermore, animal studies will be conducted to investigate the involvement of H. pylori LPS in host-pathogen interactions and its contribution to disease. This work will provide us information about the structure-function relationship of the LPS biosynthetic enzymes and their association with H. pylori pathogenicity. The knowledge acquired represents an important step in developing novel antimicrobial agents against H. pylori or as a basis for structure-guided drug discovery.