Light Microscopy - Beyond the limits of optical resolution
Michael A. Ferenczi
National Institute for Medical Research
Optics textbooks tell us that the maximum magnification obtained through a
light microscope is 400x: the closest two distinct points can be and still be
resolved is 0.2 mm. This limitation is the result of
light being diffracted by the object under observation and because diffracted
light interferes with the image.
|

|
The smallest distance between two points which
can be resolved with an oil immersion objective of the highest quality
(Numerical Aperture of 1.4) and white light was studied by Abbe (left)
in Jena in the 19th century. The resolving power of the
light microscope is empirically given by
s = l
/(2nsin
i)
where n is the refractive index of the oil, l is the wavelength of the light and i is half
the angle subtended by the objective front lens at the object plane.
Usually nsin i is called the numerical
aperture (NA) of the objective.
|
It can be seen from the formula that the resolving power improves with
shorter wavelength. Ultra violet microscopes have higher resolving power, and
the electron microscope higher still, but the latter suffers from the
inconvenience that the electron beam requires a vacuum. In spite of these
limitations, modern techniques have enabled the observation of objects smaller
than the above theoretical considerations would allow. This progress has largely
been the result of advances in fluorescence
microscopy.
Fluorescence microscopy.
Here, fluorescent molecules (fluorophores) in the object under study are visualised.
This is achieved by illuminating the object with monochromatic light at the
wavelength which excites fluorescence emission in the fluorescent molecules.
The object is viewed by means of filters which exclude the illuminating wavelength,
and select the wavelength of the light emitted by the fluorophores. Each fluorophore
can be thought of as a light source. This means that diffraction limitation
is not reached when a single fluorophore is imaged. At a sufficiently low density
of fluorophores, single molecules can be imaged. The disadvantage of the technique
is that suitable fluorophores have to be introduced into the sample. This may
be difficult, and the fluorophores may modify the behaviour of the object under
study.
In practice, many molecules have been designed to be introduced into living
cells and which bind to specific regions of the cells, highlighting the distribution
of cell compartments and even the localisation of individual proteins. Using
several different fluorophores with different optical properties, images are
obtained in living cells where different organelles appear in different colours.
The most convenient approach to obtain fluorescent images is to use an epi-fluorescence
microscope, where the illuminating light illuminates the object through
the objective. A filter block installed in the microscope tube directs the excitation
light to the objective and filters the light emitted by fluorescence in the
object and collected by the objective.
Nowadays, these microscopes are often linked to video cameras and the images
are stored digitally for electronic display and analysis. Considerable progress
has also been made in the synthesis of fluorescent molecules with improved qualities.
Factors of concern are the suitable wavelength for
excitation and emission (light should not be absorbed too much by
the cell itself), the quantum yield (how many photons of light are emitted for
a given number of photons absorbed), the bleaching
rate (in an intense beam of light, the fluorophores gradually lose
their ability to fluoresce), water solubility
and the chemistry and specificity of attachment of
the fluorophore to cellular sites. Also, the image which is observed
comes from a rather thin section of the object determined by the depth
of field (dfield) as follows:
dfield = l
/NA2 = 0.3 mm
but the fluorophores which contribute to the light collected by the objective
are molecules excited by the light in the whole cone of light exiting from the
objective. This means that the fluorescent image is superimposed on a bright
diffuse background, and may have poor contrast or even be useless unless special
precautions are taken.
Confocal microscopy
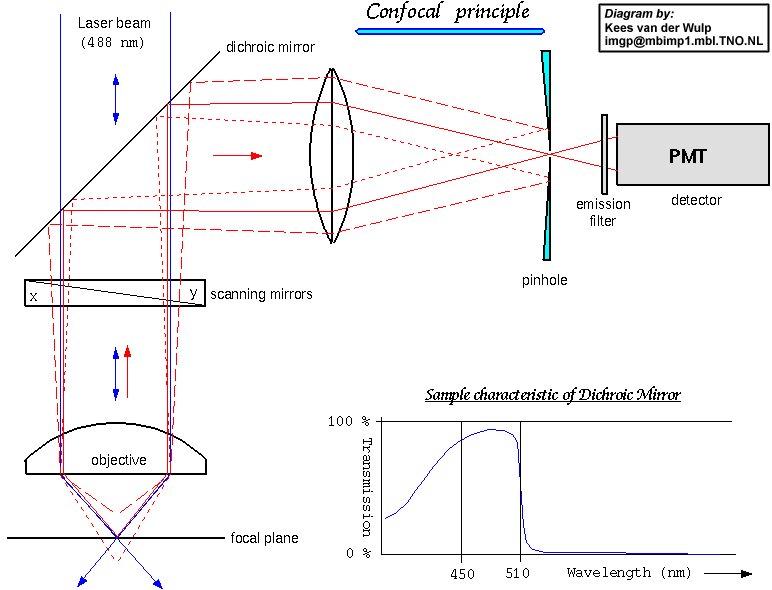
A new technique was devised to overcome the problem of high background fluorescence:
confocal laser scanning microscopy. The technique is based on an epi-fluorescence
microscope where the illumination light is provided by a laser whose beam is
expanded to fill the objective. The fluorescent light emitted by the object
is collected by the objective and focused by a secondary lens in front of a
photomultiplier tube which is used to collect the light signal. A pin-hole is
placed at the focal spot of the secondary lens. This ensures that only light
in the focal plane enters the pin-hole, and light originating from above and
below the focal plane of the object is rejected by the pinhole. The light through
the pin-hole corresponds to one spot on the object plane. To build up an image,
the pin-hole, or more usually a set of mirrors, are scanned in the x and y directions,
thereby digitally building up an image of the whole image plane. The light originates
from a narrow thickness (usually a few micrometers) of the specimen, as set
by the size of the pinhole. The digital images can then be manipulated and stunning
three-dimensional walk-through images can be produced. [see
graphical illustration]
A more detailed explanation can be found on http://www.cs.ubc.ca/spider/ladic/intro.html.
The technique however does have problems. For example the amount of exposure
of the sample to high intensity light which is required to build up an image
is considerable, resulting in bleaching of the fluorophore. The technique is
now used widely, and is constantly being refined and improved.
Total internal reflection microscopy
This technique reduces the excessive background fluorescence which originates
from fluorescent molecules which are not in the area of interest, but which
deteriorate the contrast of the image. 'Total Internal Reflection' (TIR) is
what happens to a beam of light travelling through a dense medium such as glass,
and encountering an interface with a less dense medium such as air or water.
If the beam of light encounters the surface at a sufficiently glancing angle,
the light beam will not be refracted through the interface and continue travelling
through the less dense medium, but will be reflected at the interface and continue
travelling into the glass. The angle where the transition between refraction
and total internal reflection occurs is called the critical
angle, and is given by f, the angle between
the light beam and the normal to the interface, so that:
f =
sin-1(nwater/nglass)
which for glass and water gives a value of 60.9 ¢X. At angles slightly greater
than the critical angle, namely when TIR takes place, an electric field
component of the light penetrates through the interface into the water. This
electric field is light with unusual properties. This light, the 'evanescent
wave' or 'evanescent field' has the same wavelength as the incident beam but has
the unusual property of penetrating only a short distance into the water, no
more than 1 micrometer. This means that fluorophores may be excited by the light
in the evanescent field if they are close to the glass/water interface, but
fluorophores further away in the bulk of the solution will not be excited. The
result is that images with very low background fluorescence are obtained.
 |
The image shows a myofibril obtained from a
skeletal muscle, lying on the surface of a glass prism, in the evanescent
field generated by a totally internally reflected laser beam of 532 nm.
The myofibril lies in solution containing a low concentration of Cy3-ATP,
an analogue of ATP, the substrate for the active site of the myosin
molecule. Myosin molecules are arranged into filament arrays which
interdigitate with arrays of actin molecules. This gives rise to the
striated appearance of the myofibril. The image was taken by Ronnie Burns,
NIMR, 1997. |
The image above has a high contrast, and the advantage of this technique over
confocal scanning is that the exposure to excitation light is slight, and images
can be collected very rapidly. The technique is highly suited to measuring
rapidly changing or rapidly moving objects. The technique can be used to observe
the behaviour of single fluorophores as shown below:
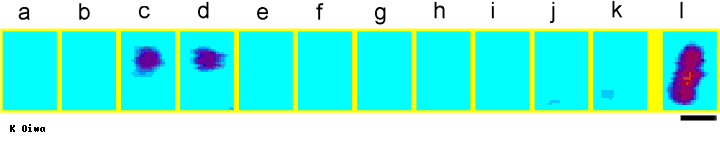
Pseudocolor display of fluorescent spots representing 50 pM
Cy3-EDA-nucleotide observed on a synthetic filament of rabbit skeletal muscle
myosin. Myosin filaments were illuminated with 7 mW Ar-ion laser (wavelength =
514.5nm). The images were taken at 3-s interval (a-k). All images were
rolling-averaged over 8 video frames. The position of the myosin filament were
confirmed as a fluorescent image in the presence of 5 nM Cy3-EDA-ATP (panel l).
Courtesy of Dr. Kazu Oiwa, Kansai Advanced Research Center, Kobe 651-24,
Japan
The main disadvantage of this technique is that the specimen are constrained
in a thin micrometer-thick layer on the glass surface. This may be an advantage
for observing the cell membrane in contact with the glass surface, but is not
suitable for observing a cell in situ.
Two-photon excitation fluorescence microscopy
A further technique has been developed to achieve observation of cells in
their tissue. This technique makes use of the fact that fluorescence can be
excited in fluorophores not only by photons at a wavelength corresponding to the
absorption ban for fluorescence, but also by photons at a wavelength twice that
of the absorption band. The latter absorption only produces fluorescence if two
photons are aborbed in quick succession, namely within a few femtoseconds of
each other. The consequence of this two-photon excitation technique is that
fluorescence emission only takes place where the photon density I sufficient to
meet the photon-flux requirements for two-photon excitation, namely at the focal
point of a microscope objective through which light from a high repetition rate
laser travels. The consequence is that at any one time, the fluorescence
originates in a small volume of the object. Scanning in the x-y plane and
through the depth of the objects allows 3-D reconstruction of single cells as
shown below, in a living tissue.
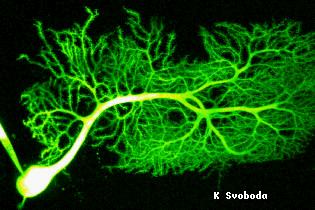 |
Purkinje neurone in a living brain slice filled
with fluorescein dextran imaged with two-photon excitation laser scanning
microscopy. Size of the image is 200x160 microns. By courtesy of Dr. Karel
Svoboda, Cold spring Harbor Laboratories, Cold Spring Harbor, NY 11724,
USA. |
The Future
We have briefly seen that developments in techniques have led to images which
would have been undreamt of, only a few years ago. Progress on many fronts was
necessary: there has been new chemistry for developing biologically useful fluorophores,
there has been considerable progress in camera technology, to record as many
of the available photons as possible. Computing development has enabled scanning
techniques to be affordable in many laboratories, and computer-aided lens design,
and new lens materials, have produced lenses of higher quality and numerical
aperture than ever. There is little reason to doubt that this progress will
continue, and single molecule imaging will become routine. New techniques are
also developing. Perhaps the most significant one is Atomic
Force Microscopy where again the driving force is the interface between
physics and biology - and that is Biophysics.
[Electron Microscopy]
{kind=link}