|
INTRODUCTION |
Transposition is a process in which a defined DNA sequence, called a
transposable element, moves from one location to a second location
on the same or another chromosome. Transposable elements occur
widely in nature and include the simple insertion sequences or
composite transposons of bacteria, certain bacteriophages, transposons,
and retrotransposons of eukaryotic cells and retroviruses such
as HIV-1.1 Originally described by McClintock (1)
in a series of elegant experiments of controlling elements in
maize, transposons have been found in all phyla studied to date,
including humans. These mobile genetic elements are likely to
have played a role in genome evolution and continue to shuffle
antibiotic resistance traits among bacteria today (for a general
review, see Ref. 2). In eukaryotic species,
transposons
are not only numerous but also very promiscuous and are known
to cause chromosome mutations. Also, the DNA cleavage reactions
involved in immunoglobulin gene rearrangement have been shown
to occur via a transposition mechanism (3).
Achieving a molecular and structural understanding of transposition
has been a formidable challenge in part because of the complexity
of the process. Transposition is initiated by the binding of
a transposable element-encoded protein called a transposase to
specific DNA sequences located at or near the ends of the element.Next,
the DNA-bound transposase oligomerizes to form a synapticnucleoprotein
complex. Thereafter cleavage of one or both strandsat the transposon ends
occurs where the exact cleavage sites area property of the specific element
(4, 5). The initial strand cleavage
reaction is believed to occur via nucleophilic attack of an
activated water molecule on the phosphodiester bond at the end
of each element to leave a 3' OH group. As described below, IS4
family elements, such as Tn5 and Tn10, have a more complexmechanism
in which formation and cleavage of a hairpin intermediateleads to 5' end
release. In the final step, the 3' OH performsa nucleophilic attack on
the target DNA, leading to strand transfer.
It has proved troublesome to study the structural properties of these
enzymes since it has been difficult to crystallize a full-length
protein for any of the transposases or the integrases due to
their poor solubility properties (6, 7).
This problem might be attributable to the apparent structural
flexibility introduced by the presence of distinct modules responsible
for the DNA binding and catalytic activities. As a consequence
studies have focused on isolated domains that are responsible
for part of the function of the protein. This approach has yielded
the three-dimensional structures for the catalytic core domains
of Mu transposase, and HIV-1 and avian sarcoma virus (ASV) integrases
(8-10). These fragments contain that part
of the intact molecule responsible for the 3' strand cleavage
and transfer reactions, which are both phosphoryl transfer reactions.
This has been demonstrated for the truncated forms of HIV integrase
and ASV integrase proteins that have been found to retain the
ability to perform a "disintegration" reaction that mimics the
reverse of the strand transfer step (11-13). Remarkablythese
catalytic domains exhibit a common fold that appears tobe related to a
broader class of polynucleotidyltransferases thatincludes RNase H, both
from Escherichia coli and HIV-1 reversetranscriptase, and recombination
factor RuvC (14-18). This has led to speculation
that the catalytic mechanism of the transposase/integrase superfamily
may be similar to the exonucleolytic cleavage reaction of E.
coli DNA polymerase I (17).
The catalytic core domains of the Mu and HIV-1/ASV transposase/integrase
enzymes consist of a central five-stranded mixed parallel and
antiparallel  -sheet sandwiched between
four
-sheet sandwiched between
four -helices. This fold brings
three essential carboxylate residues, two aspartates and one
glutamate, into close proximity at a shallow cleft on one surface
of the protein. These acidic residues are common to all transposases
and form the "DDE" motif believed to be responsible for coordinating
the divalent metal ions necessary for catalysis. In the case
of RNase H of HIV-1 and ASV integrase, a pair of divalent cations
has been observed, coordinated by the three conserved carboxylates
(19, 20). A magnesium ion has also
been observed within the active site of HIV-1 integrase (21).
Although the structures of the individual core domains have
proved to be of immense value for understanding this family
of proteins, the relationship between the functional segments
is lost by the strategy of divide-and-conquer. For example,
these structures do not provide information about the possible
locations of the DNA binding domains, nor do they show how different
domains interact with one another. Thus, to understand transposition
in more complete detail, we have undertaken a multidomain structural
study of Tn5 transposase.
-helices. This fold brings
three essential carboxylate residues, two aspartates and one
glutamate, into close proximity at a shallow cleft on one surface
of the protein. These acidic residues are common to all transposases
and form the "DDE" motif believed to be responsible for coordinating
the divalent metal ions necessary for catalysis. In the case
of RNase H of HIV-1 and ASV integrase, a pair of divalent cations
has been observed, coordinated by the three conserved carboxylates
(19, 20). A magnesium ion has also
been observed within the active site of HIV-1 integrase (21).
Although the structures of the individual core domains have
proved to be of immense value for understanding this family
of proteins, the relationship between the functional segments
is lost by the strategy of divide-and-conquer. For example,
these structures do not provide information about the possible
locations of the DNA binding domains, nor do they show how different
domains interact with one another. Thus, to understand transposition
in more complete detail, we have undertaken a multidomain structural
study of Tn5 transposase.
Besides providing a broader context for understanding transposition
in general, structural information about Tn5 transposase has
the potential to provide specific understanding of the IS4 family.
Two representative IS4 family transposases, those encoded by
Tn5 and Tn10, have been the object of extensive genetic studies(for
reviews see Refs. 22 and 23). The
literature on these elements provides a detailed knowledge base
by which to interpret the structure of Tn5 and will allow
this structure to serve as a basis for future structure/function
analyses. Primary sequence examination of the IS4 transposase
family suggests that, although they undoubtedly contain DDE
residues functioning in divalent metal coordination, the locations
of these residues are placed differently in the primary sequence
than those found in retroviral integrases or MuA. In addition,
comparison of IS4 transposase primary sequences and genetic
studies with Tn10 transposase (24, 25)
and Tn5 transposase2 suggests that
the IS4 transposases contain some critical motifs (such as the
Y(2)R(3)E(6)K motif discussed below) not found in other transposases.
Finally, IS4 transposases catalyze two additional phosphoryl
transfer reactions, in comparison with retroviral integrases and
MuA transposase, to generate blunt-ended transposon DNA as opposed
to only nicking the DNA. In these IS4 elements the 3' OH group
formed by the initial strand cleavage reaction attacks the complementary
strand to cleave the element from the donor DNA leaving a hairpin
intermediate (27).3 Presumably,
this hairpin intermediate is cleaved by the attack of a second
water molecule to expose the 3' OH group and leave a blunt end.
The resultant 3' OH acts as a nucleophile in the subsequent
end strand transfer reaction by attacking a phosphodiester bond
on the target DNA. As is the case with the reactions of retroviralintegrases
and Mu transposase, these reactions require only divalentcations as cofactors.
Understanding the structure of the Tn5 protein will provide
a basis for understanding these unique features of the IS4 family
of transposases.
Tn5 is a composite transposable element found in Gram-negative
bacteria and consists of two IS50 insertion sequences thatflank,
in inverted orientation, three genes encoding antibioticresistances (for
reviews of Tn5, see Refs. 22 and
28).
Each IS50 is bordered by two related 19-base pair sequences,
the outside end (OE) and the inside end. IS50R encodes
the 476-amino acid transposase. Purified transposase has been
found to be necessary and sufficient for catalysis of Tn5
transposition in vitro in the presence of pairs of OE
DNA ends and Mg2+ (29). Transposase releases
the transposon from donor DNA leaving blunt ends and inserts
it into a 9-base pair staggered cut site in target DNA (30,
31).
A closely related transposase, Tn10,is thought to form synaptic
complexes in which a monomer is responsiblefor all of the catalytic events
at each transposon end (25).In contrast, Mu transposase
has been shown to function as a tetramerwith a dimer at each mobile element
end (32,
33). Complementationstudies
of HIV-1 integrase mutants have suggested that this enzymealso acts as
a dimer at each viral end (34).
In E. coli, transposition levels must be tightly regulated in
order to prevent excessive chromosome mutagenesis. Tn5 employsa
unique means of self-control by expressing a truncated versionof the transposase
that functions as an inhibitor. This inhibitorprotein contains 421 amino
acid residues and differs from thefull-length transposase only by the absence
of the first 55 N-terminalamino acid residues. The inhibitor utilizes a
distinct initiationsite relative to the transposase. In vivo, the
inhibitor proteinis a natural transdominant negative regulator of transpositionand
acts presumably by forming inactive mixed multimers with transposase,not
by competitive DNA binding (35). Interestingly, transposaseitself
can act as an inhibitor when present at sufficient levels(36,
37).
Many obvious questions remain concerning the molecular basis of Tn5
transposition. In particular what are the protein-protein interactions
that occur within the synaptic complex? How are the catalytic
centers related to each other in the synaptic complex? What
is the structure of the catalytic core? How does the non-productivemultimerization
occur? In an effort to answer these questionswe have determined the structure
of the intact IS50 Tn5 inhibitor. This protein
represents 88% of the full-length transposase sequence. This
is the first structure of a naturally expressed biologically active
transposase fragment and is the most complete transposase structure
known. On the basis of its sequence similarity to other transposases,
this protein is predicted to contain all of the critical catalytic
core regions of the full-length transposase (24)
and has been shown to contain the determinants for dimerization (35,
38).
Proteolytic studies also suggest that Tn5 inhibitor has
a tertiary structure that is similar to full-length transposase (38).
The inhibitor structure reveals that the catalytic domain of Tn5
transposase shares similar structural features with those of
HIV-1, ASV, and Mu transposase/integrase even though they share very
low sequence homology, although it does include an additional extended -sheet.
It also confirms the presence of the DDE catalytic motif of
the superfamily and reveals the location of an arginine residue
in the active site that is strictly conserved in the IS4 subfamily
of transposases. The structure suggests that the catalytic motif
is "DDRE" for this group of enzymes. This study extends the
common framework for transposition to prokaryotic insertion sequences.
The Tn5 inhibitor is dimeric where the interface occurs in
the C-terminal region of the protein and is dominated by the interaction
of two helices that form a scissor-like interaction. Together,
these observations provide insights into catalysis and suggest
models for the structural basis of regulation of transposition and
for the nucleoprotein architecture within transposition intermediates.
|
EXPERIMENTAL
PROCEDURES |
Protein Purification, Crystallization, and X-ray Data Collection--
The inhibitor protein was prepared and purified as described previously
(38, 39). In the final step of the
purification, the protein was eluted with a salt gradient from
a DEAE-anion exchange column. The pooled fractions containing
the inhibitor protein were concentrated to ~16 mg/ml and dialyzed
against 100 mM tetraethylammonium sulfate
and 20 mM Tris at pH 7.9. The inhibitor protein
was crystallized at room temperature by micro batch. Typically 15
?l of protein at 16 mg/ml was combined with an equal volume of
20% PEG 8000, 100 mM tetraethylammonium sulfate, and
100 mM MES, pH 6.0. Crystals grew spontaneously
or were micro-seeded and reached a size of 0.7 × 0.4 × 0.3
mm in 14-28 days. Precession photography determined that the
crystals belong to the space group P21212.
Unit cell parameters are a = 182.4 ?, b = 72.6 ?, andc
= 41.7 ? for native crystals measured at  6
°C, and a = 181.8 ?, b = 71.9 ?, and
c
= 41.3 ? for the platinum derivative recorded at160
°C with synchrotron radiation. There is one molecule per asymmetric
unit and a solvent content of 57%. Crystals for preparation of
heavy atom derivatives and data collection on the laboratory area
detector were stabilized in a synthetic mother liquor containing 19%
PEG 8000, 100 mM tetraethylammonium sulfate, 300 mM
NaCl, and 50 mM MES, pH 6.0. Crystals used
for data collection with synchrotron radiation were transferred
sequentially into a cryoprotectant solution containing 19% PEG
8000, 100 mM tetraethylammonium sulfate, 300
mM NaCl, 50 mM MES, pH 6.0, and
15% ethylene glycol and flash-cooled to approximately 160
°C in a nitrogen stream (40,
41).
6
°C, and a = 181.8 ?, b = 71.9 ?, and
c
= 41.3 ? for the platinum derivative recorded at160
°C with synchrotron radiation. There is one molecule per asymmetric
unit and a solvent content of 57%. Crystals for preparation of
heavy atom derivatives and data collection on the laboratory area
detector were stabilized in a synthetic mother liquor containing 19%
PEG 8000, 100 mM tetraethylammonium sulfate, 300 mM
NaCl, and 50 mM MES, pH 6.0. Crystals used
for data collection with synchrotron radiation were transferred
sequentially into a cryoprotectant solution containing 19% PEG
8000, 100 mM tetraethylammonium sulfate, 300
mM NaCl, 50 mM MES, pH 6.0, and
15% ethylene glycol and flash-cooled to approximately 160
°C in a nitrogen stream (40,
41).
Initial native data and all heavy atom derivative data for MIR phasing
were collected to 2.9-3.5-? resolution at 6
°C with a Siemens HiStar area detector at a crystal to detector
distance of 18 cm. CuK radiation
was generated by a Rigaku RU2000 rotating anode x-ray generator
operated at 50 kV and 90 mA and equipped with Siemens G?bel
mirrors. Diffraction data frames of width 0.15° were recorded
for 90-120 s. The frames were processed with XDS (42,
43)
and internally scaled with XCALIBRE.4 Tables
I-III
display the diffraction data sta tistics for the native, heavy
atom derivative, and MAD phasing data sets.
Crystallographic Structure Determination-- A structure of the
Tn5 inhibitor protein was initially determined by multiple isomorphous
replacement from five heavy atom derivatives and subsequently
confirmed by multiple wavelength anomalous dispersion from one
heavy atom derivative (45) (Tables I-III).
Derivatives were prepared by soaking crystals in a solution of
synthetic mother liquor containing one of the following: 0.5 mM
MeHgCl, 1 mM Au(CN)2, 1 mM
ter(pyridine)PtCl, 0.5 mM di-|mu|-iodobis(ethylenediamine)di-platinum(II)
nitrate (PIP), or 1 mM bis(pyridine)PtCl. The heavy
atom positions were determined from difference Patterson maps
and placed on a common origin with difference Fourier maps.
The occupancies and positions of the heavy atom binding sites
were refined with the program HEAVY (46).
The initial phases were modified by solvent flattening with
the algorithm of Kabsch and co-workers (48)
and utilized to improve the heavy atom refinement (47,48).
Phase calculation statistics for these derivatives are included
in Table I. A polyalanine model was built into the subsequentelectron
density map with the software package FRODO (49,
50).In
the early stages of model building, the heavy atom phases werecombined
with model phases with SIGMAA weighting (51). Thereafterthe
model was improved through cycles of manual model buildingand least squares
refinement with the program TNT (52). The crystallographic
R-factor
for the model refined against the data collected at 6
°C was 22.1% for all data measured from 30 to 2.9 ?.
In order to confirm the validity of the structure of the Tn5
inhibitor protein, additional independent phasing information was
obtained from multiple wavelength anomalous dispersion (MAD) measurements.
MAD data were collected from a single crystal soaked in 1 mM
ter(pyridine)Pt (II) for 12 h. The x-ray wavelengths were chosen
from the x-ray fluorescence spectra of the platinum L-III edge
recorded directly from the crystal in order to optimize the anomalous
dispersion effects from the platinum atoms. The MAD data were
recorded with a 3 × 3 tiled CCD detector on the insertion device
on beam-line 19 of the Structural Biology Center at the Advanced
Photon Source in Argonne, IL. The crystal to detector distance
was 260 mm, and the data were collected with frames of width
1.5°. Diffraction data were processed using the HKL 2000 software
package (53, 54). The Friedel differences
in the reference data set ( = 1.0273 ?) were externally local scaled to remove systematic
errors. Thereafter the other three data sets were placed on
a common scale by local scaling to the reference data set (55).
This strategy had a profound effect on the quality of the subsequent
electron density map. Phases from the MAD data sets were calculated
with the program SOLVE (46, 56) and
improved by solvent flattening with the program DM (57,
58).
The model of Tn5 inhibitor protein based on the MIR phases
was oriented into solvent-flattened map with the program AMORE
(59). Visual inspection of the map showed
that the tracing of the-carbonbackbone
in the initial MIR structure was correct. The electrondensity map was improved
by combining MAD phases with model phaseswith SIGMAA weighting (51).
A portion of representative electron density is shown in Fig.
1.
Thereafter the model was improved through cycles of manual model
building and least squares refinement with the program TNT (52).
The final structure has a crystallographicR-factor of 19.5% at a
resolution of 2.9 ?. Refinement statisticsare listed in Table
IV.
= 1.0273 ?) were externally local scaled to remove systematic
errors. Thereafter the other three data sets were placed on
a common scale by local scaling to the reference data set (55).
This strategy had a profound effect on the quality of the subsequent
electron density map. Phases from the MAD data sets were calculated
with the program SOLVE (46, 56) and
improved by solvent flattening with the program DM (57,
58).
The model of Tn5 inhibitor protein based on the MIR phases
was oriented into solvent-flattened map with the program AMORE
(59). Visual inspection of the map showed
that the tracing of the-carbonbackbone
in the initial MIR structure was correct. The electrondensity map was improved
by combining MAD phases with model phaseswith SIGMAA weighting (51).
A portion of representative electron density is shown in Fig.
1.
Thereafter the model was improved through cycles of manual model
building and least squares refinement with the program TNT (52).
The final structure has a crystallographicR-factor of 19.5% at a
resolution of 2.9 ?. Refinement statisticsare listed in Table
IV.
View larger version (79K):
[in
this window]
[in
a new window] |
Fig. 1.
Stereo view of the electron density associated with central -sheet
that forms the base of the predicted divalent cation-binding site.
The electron density was calculated with coefficients of the form 2FoFc
and displayed with the program MOLDED (69) and MOLSCRIPT
(70). |
|
|
RESULTS |
Overall Structure-- The inhibitor protein contains 421 amino
acid residues and corresponds exactly to residues Met56-Ile476
of the full-length transposase. Even though the inhibitor proteinis expressed
independently from its own initiation site, and thusis a protein in its
own right, the residue numbering utilizedin this paper will be that of
the corresponding amino acids inthe Tn5 transposase. The current
model for the inhibitor startsat Ser70 and terminates at Gln472.
Although much of the structure is well defined, many of the loops
exhibit considerable flexibility. This flexibility gives rise
to breaks in the electron density between Arg104-Trp124,
Val246-Arg256, and Met343-Pro346.
In addition to these breaks in the polypeptide chain, the followingamino
acids were disordered beyond the -carbon:
Glu72, Glu88, Asp133, Arg215,
Lys216, Lys244, Val246, Gln341,
Arg342, Met343, Pro346, Asp347,
Asn348, Leu349, Met352, Asp400,
and Glu417.
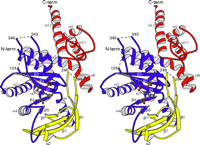
The structure of the inhibitor protein may be divided into two major
domains as shown in Fig. 2, a catalytic domain and aC-terminal
dimerization domain. Residues Ser70-Gln365 form the
catalytic domain. This region is a mixed /
structure and contains the carboxylate residues that have been
implicated in metal binding. The catalytic domain is built from
seven -helices and nine
strands of mixed parallel-antiparallel -sheet.
The first five strands of sheet and four of the helices bear
striking structural similarity to the HIV-1 integrase, ASV integrase,
and Mu transposase cores, as well as to RuvC and RNase H of
HIV-1 (also RNase H from E. coli) as discussed below.
Residues Arg104 to Trp124 and Leu224 to
Leu309 represent insertions relative to the core structures
of the other integrases. The first insertion includes a 20-residue
disordered loop located between 1
and 2. The insertion from Leu224
to Leu309 occurs between 5
and 6 and serves to increase the
breadth of the sheet from five to nine strands and to deepen
the active site cleft. A long-helix, 6,
extending from Leu309 to Gly335 lies across the face
of the -sheet and contributes to the
structural foundation of the active site. The hydrogen bonding
pattern in this helix is disrupted near the active site between
residues 320 and 324. The final secondary structural element
in the catalytic domain is helix 7,
which extends from Glu350 to Ala378. This helix couples
the catalytic domain to the C-terminal dimerization domain.
There is a prominent bend in this helix at Leu366, and this
is taken as the dividing line between the two domains.
The C-terminal domain (residues Leu366-Gln472)
contains five -helices (7
to 11) and is responsible for the
dimer interface observed in the crystal lattice (Fig.
3).
It is an extended domain that conveys the impression that this
component of the structure has the potential for flexibility.
Helices 9 and 11 form extensive interactions with a neighboring
molecule across the crystallographic dyad axis as discussed
below.
View larger version (73K):
[in
this window]
[in
a new window] |
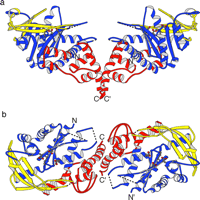
Fig. 3.
Ribbon representation of the dimer viewed perpendicular to the 2-fold
axis (a) and along the crystallographic 2-fold axis (b).
The color scheme is as follows: blue, the structurally conserved
catalytic core amino acid residues Ser70-Leu224,
Leu309-Gln365;
yellow, -sheet
insertion, Leu224-Leu309;
red, C-terminal
dimerization domain, Leu366-Gln472. The active site
residues, Asp97, Asp188, Arg322, and Glu326,
are included in
ball-and-stick representation. |
|
The structure is consistent with results obtained from partial proteolysis
of Tn5 transposase and the inhibitor protein where many
of the cleavage sites coincide with surface loops. The N-terminalregions
of both proteins appear to be susceptible to proteolysiswith proteolytic
sites after Arg61 and Lys113 (38).
Lys113 coincides with a disordered segment of the inhibitor
structure. The major proteolytic cleavage region, residues Lys252-Leu263,
corresponds to the flexible loop that contains the disordered residues
Val246-Arg256 (38). Likewise,
the proteolytic region bounded by residues412-440 is located within the
extended C-terminal domain and isrelatively solvent-exposed which accounts
for the proteolyticsensitivity. It is noteworthy that the tryptic digestion
patternsand cleavage sites of the Tn5 transposase and the inhibitor
proteins are very similar which suggests that both proteins
contain the same fold.
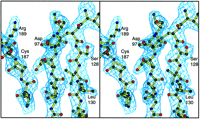
The Active Site-- Inspection of the Tn5 inhibitor protein
structure reveals that three carboxylate residues (Asp97, Asp188,
and Glu326) reside in close proximity to one another and are
associated with a basic residue, Arg322 (Fig. 4).
The three residues map close to the position of the catalytic
triad in the ASV integrase structure and correspond to the characteristic
DDE motif described for transposases of the IS3 family, for
Mu transposase and for the retroelement integrases as well as
for the mariner/Tc3 family of eukaryotic transposases (24,
60,
61).
Changing Glu326 to alanine results in loss of catalytic activity
of Tn5 transposasein vivo.2 Sequence alignment
with Tn10 transposase based on an N-terminalregion of homology (38)
and a C-terminal extended region of homology called C1 (24)
shows that Asp97, Asp188, Glu326, and
Arg322 of Tn5 transposase correspond to four conserved
residues of Tn10which have been shown to be required for catalytic
activity (25).The arginine is strictly conserved throughout
the IS4 family (24).Thus the structure of the Tn5
transposase active site confirmsthe presence of the DDE carboxylate cluster
and suggests thatthe catalytic motif for the IS4 family should be expanded
to DDRE.

View larger version (44K):
[in
this window]
[in
a new window] |
Fig. 4.
Stereo close up view of the active site carboxylate residues and the
associated Y(2)R(3)E(6)K motif. The conserved carboxylates, Asp97,
Asp188, and Glu326 in Tn5 inhibitor protein
are compared with the equivalent residues in the ASV integrase core structure
(PDB accession number 1VSD,
Ref. 44). The inhibitor is depicted in ribbon and ball-and-stick
representation, whereas active site residues for ASV integrase are colored
in
green. |
|
The presence of the arginine side chain prevents the three carboxylate
groups from coming as close together as they do in the ASV integrase
structure. Unless the side chain of Arg322 undergoes a major
conformational change upon binding of divalent metal ion(s)
and/or substrate, it is difficult to foresee how the transposase
active site could be made to resemble exactly the ASV integrase
active site, in terms of its coordination of metal ions. The
function of arginine in transposase might be to partially neutralize
the negative charge on the acidic residues or to orient the
carboxylate groups so that they might support a more open coordination
for the divalent cations.
Dimer Interface-- The C-terminal dimerization domain of the Tn5
inhibitor protein observed here has no analog in any of the previously
published transposase/integrase structures. This domain contributes
to the interface between two molecules across a crystallographic
2-fold axis that is formed by -helices
9 and 11. The long C-terminal helices of adjacent molecules
pack against one another from residues Ser458 to
Met470 at an angle of 65°. Interestingly the C-terminal helices
come in very close contact. This is facilitated by the presence
of Gly462 at the crossover point which allows for
a separation of only 3.9 ? between adjacent -carbons.
Helix 9 is nearly perpendicular to the C-terminal helix, and
it makes contacts with the C-terminal helix,but not with its
counterpart on the symmetry-related molecule. The subunit-subunit
interactions are primarily hydrophobic in nature and bury approximately
700 ?2 of solvent-accessible surface area. This modest interaction
most likely represents the homodimer interface in the inhibitor
protein and may account for the facile interchange between monomers
and dimers in solution (62).
|
DISCUSSION |
Comparison of Transposase/Integrase Catalytic Domains-- One of
the most remarkable features of the retroviral integrases and Mu transposase
is the observation that, even with very low sequence similarity,
a significant degree of secondary and tertiary structure conservation
exists between their catalytic domains. Even the functionally
divergent proteins RNaseH and RuvC exhibit a similar fold. The
common core observed in these integrases and transposases consists
of five -strands laid out in a threeparallel/three
antiparallel configuration sandwiched between fourconserved -helices.
This fold forms a shallow groove with the catalytic acidic residues
located at its base. The first and fourth-strands
contribute the two aspartate residues, and a helix near the
C terminus of the catalytic core domain (or coil, in the cases of
Mu transposase and HIV-1 integrase structures) contributes the
glutamate residue. Given the previously observed structural similarity
between these enzymes, it is not surprising that Tn5 transposase
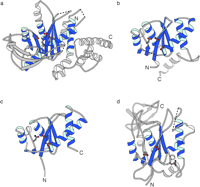
inhibitor contains a similar folding motif as shown in Fig.
5.
For example the r.m.s. difference between the coordinatesfor 81 structurally
equivalent -carbons in Mu transposase
and the inhibitor protein is 1.77 ? even though the overall
sequence identity for these residues is 9%. A numerical comparison
between the Tn5 inhibitor protein and the core structures
of retroviral integrases and Mu transposase is given in Table
V.
View larger version (52K):
[in
this window]
[in
a new window] |
Fig. 5.
Structural comparison between Tn5 inhibitor protein (a),
HIV-1 integrase (b), ASV integrase (c), and Mu transposase
proteins (d). The structural features common to all of these proteins
are colored in blue. The structures were aligned on the core of
the Tn5 inhibitor protein with the program OVRLAP (71).
The coordinates for the ASV and HIV-1 integrases, and MU transposase core
structures were obtained from the Brookhaven Protein Data Bank (accession
numbers 1VSD,
1BIU,
and
1ITG,
respectively (10, 21, 26,
44)). |
|
View this table:
[in
this window]
[in
a new window] |
Table V
Structural comparisons between the Tn5 inhibitor
protein and the core structures for Mu transposase and the retroviral integrases
|
|
There are, however, two insertions that distinguish the Tn5 transposase
catalytic domain from the previously reported structures for
Mu transposase and HIV-1/ASV integrases. The first of these is
a large partially disordered 24-residue loop (Leu101-Trp124)
between 1 and 2.
The corresponding loop varies from two residues in HIV-1 integrase
to 15 residues in Mu transposase. In the previous structures,
the loop between 1 and 2
is ordered in the crystal structure. It is possible that the
large disordered region of this loop in the Tn5 transposase
only becomes ordered upon binding to DNA. Interestingly, this
loop is located near the active site and also near the N terminus
of the inhibitor protein. In the full-length protein, such an
arrangement positions this loop between the site of DNA cleavage
and the presumed location of the N-terminal DNA binding domain.
It is therefore conceivable that this loop may help orient the
transposon DNA in the active site for catalysis.
There is also an insertion of 86 amino acid residues (Leu224-Leu309),
relative to ASV and HIV-1 integrase and Mu transposase, between the
conserved fifth -strand and the -helix
that carries the conserved catalytic glutamic acid residue.
This insertion is mostly-strand where the
additional residues serve to increase the breadth of the -sheet
by adding four more antiparallel strands at one edge (Figs.
2
and 3). As a consequence of the curvature of the-sheet,
these additional strands wrap around the long -helix,6,
that forms the foundation of the active site and forms a distinctwall that
overlooks the catalytic carboxylates. These additionalstructural elements
change the active site from a shallow depressionobserved in the Mu transposase
and retroviral integrase structuresto an elongated canyon in the Tn5
protein. Although the functionof the insertion in Tn5 is unknown,
it is interesting that the partially disordered loop (Ile241-Lys260)
that lies at the edge of the inserted sheet contains eight positivelycharged
residues and suggests that these might contribute to thenonspecific DNA
binding component of the transposase. The Mu transposasecontains a traditional -barrel
in addition to its catalytic domain; however, this is located
in a different position. Its subdomain is located at the C terminus
of the catalytic domain and is located on the opposite side
of the protein relative to the active site such that its function
is clearly different from the insertion in the Tn5protein.
The YREK Signature-- The catalytic arginine and glutamate residues
discussed above are part of a signature sequence, Y(2)R(3)E(6)K, characteristicof
many, but not all, transposases of the IS4 family (24).
Mutation of the corresponding Tyr to Phe in Tn10 transposase
resulted in a decrease to 83% of wild type transposition activity
in
vivo (63). In the Tn5 protein
structure, Tyr319 is partially buried adjacent to the carboxylate
group of Asp188, one of the active site aspartate residues.
Since transposition was decreased by only 17% in the tyrosine
to phenylalanine mutant of Tn10 transposase, it seems
likely that the tyrosine does not play a direct role in catalysis.
Interestingly the YS mutant in Tn10 (63)
and a YA mutant in Tn52 eliminated the enzymatic activity
which suggests that the phenyl group may be important for stabilizing
the tertiary structure of the active site. The function of the
conserved Lys of the YREK signature is less clear. Mutation
of the Lys to Ala in Tn5 transposase resulted in a mutant
that impaired cleavage.2 This result is in contrast to a mutation
of the Lys to Ala in Tn10 transposase that resulted in
a mutant that allowed cleavage but was defective in target capture
or strand transfer (25). In the inhibitor
protein structure, this residue is solvent-exposed and does
not interact with any of the active site residues. The amino group is located >10 ? away from the carboxyl group of Asp97
and resides at the base of the active site canyon in the Tn5structure.
This amino acid could be involved in retention or orientationof substrate
DNA during cleavage or strand transfer.
amino group is located >10 ? away from the carboxyl group of Asp97
and resides at the base of the active site canyon in the Tn5structure.
This amino acid could be involved in retention or orientationof substrate
DNA during cleavage or strand transfer.
Possible Interactions between N- and C-terminal Domains-- It
is clear that the protein-protein interactions involved in homodimers of
the Tn5 transposase and homodimers of the Tn5 inhibitor
protein are somewhat different since the Tn5 inhibitor protein
can homodimerize in solution under conditions where the transposase
is predominantly monomeric (35).5
Yet the proteins differ only by the presence of an additional 55
amino acids in the transposase where these amino acids unambiguouslyparticipate
in specific binding to OE DNA (64,
65).
This implies that the specific DNA binding domain of the transposase
influences dimerization. Inspection of the inhibitor structure
shows that the N terminus and C terminus of the inhibitor structure
are located near each other, and thus, presumably the N-terminal
DNA binding domain and the C-terminal dimerization domain also
lie close to one another in transposase. This suggests that
the transposase N-terminal and C-terminal domains interact in
such a way that the N-terminal domain prevents the C-terminal
domain-mediated dimerization. It should also be noted that the
C terminus of Tn5 transposase is known to inhibit N terminal-mediated
DNA binding (39,
66).
The monomeric nature of Tn5 transposase may have functional
consequences for transposition. It seems plausible that monomers
of transposase bind OE DNA ends and that synapsis of monomer-bound
ends leads to productive transposition. Inhibition appears to
occur via dead-end complexes through C-terminal heterodimerizationof a
monomer-bound end with an inhibitor molecule (67).
Significance of the Dimer Interface-- Protein-protein interactions
are important for proper nucleoprotein synaptic complex formation in all
transposases. Tn5 transposase is unique in its use of
non-productive protein-protein interactions, involving both
the inhibitor protein and transposase, to accomplish inhibition
and a related phenomenon of transposase cis-restriction in
vivo (65). The protein-protein interactions observed
in the Tn5 inhibitor structure appear to be involved
in the process of inhibition but not synapsis.
The observed dimer conformation of Tn5 inhibitor does not appear
to represent a structure that might form the basis for a model
of synapsis even though it does contain some attractive elements.
For example, inspection of the model shows the catalytic sites
are positioned on the same side of the dimer. It would be easy
to imagine a concerted strand transfer reaction; however, the
distance between the active sites in the dimer is approximately 65
?, which is too far apart to account for the 9-base pair spacing between
the cuts made in the target DNA during strand transfer. If the
observed dimerization interface is present at synapsis, then
a major domain rearrangement must take place to bring the active
sites on the two molecules of the dimer closer together. It
is not easy to predict how this might occur, but if the interactionbetween
the C-terminal domains is preserved at synapsis, a simpleway to accomplish
this might be to rotate the domains downwardand allow the catalytic domains
to approach more closely.
A plausible hypothesis is that the dimer interaction in the inhibitor
structure represents the structure of the inhibited complex.
The role of the C-terminal domain in inhibition is suggested by
a point mutation located within the long helix of the C-terminal dimerization
motif that was designed on the basis of the structure reported
here. The mutant AD466 in the inhibitor protein is observed to
prevent homodimerization of the inhibitor protein and eliminates its
inhibitory effect on transposition by presumably preventing the
formation of the transposase-inhibitor complex on DNA (67).Another
line of evidence that implicates the observed dimer interfacein inhibition
is a primary sequence alignment analysis of Tn10and Tn5 transposases
that indicates that the least conserved regions occur at the
C termini. It is of interest that Tn10 transposase is
not negatively regulated by protein dimerization but does undergosynapsis.
Therefore, the dimer interface observed in Tn5 inhibitorprotein
may have no counterpart in Tn10 (38). Thus a
role for the C-terminal dimer interface in inhibition but not
synapsis is suggested.
Constraints on the Structure of the Synaptic Complex-- Since
the protein-protein interactions in the structure are unlikely to be representative
of the synaptic complex, it is possible that formation of the
synaptic complex would involve a different dimer interaction.
Two regions of transposase have been identified as containing
determinants for dimerization based on far Western studies of
proteolytic products, residues Leu114-Arg314 and
residues Thr441-Ile476 (38). Clearly
the latter region falls within the dimerization domain observed
here. The implication of residues Leu114-Arg314 in
the dimerization by the proteolytic studies must be viewed with
caution since it is uncertain whether the isolated fragments that
implicate this region could fold into functional domains; however,
DNA binding studies have shown that the first 387 amino acids
of transposase are sufficient for dimerization (66,
68).Although
the inhibitor does not bind OE DNA specifically, it interactswith an OE-bound
transposase monomer in a ternary complex as shownin gel shift experiments
(35,
66). Interestingly completeremoval
of the dimerization domain from the transposase by truncationat residue
369 eliminates dimerization of the DNA-protein complex,whereas truncation
at 387 retains the ability for dimerization(66,
68).
These results suggest that there exists a seconddimerization region. Thus
distinct dimerization regions couldbe used for inhibition and synapsis.
Since the fold of the catalytic domain of Tn5 inhibitor protein
is similar to those of the HIV-1 and ASV integrase core domains, it
is appropriate to consider whether the Tn5 protein might dimerizein
a similar manner to those proteins. Dimer interactions observedin the crystal
lattices of HIV-1 and ASV involve interactionsof integrase helices 1
and 5 (8, 9).
The corresponding structural elements of Tn5 are helices 2
and 7. These two elements encompass
amino acid residues that fall in the range of 114-314. Although
this is an attractive proposal, it is highly unlikely that this
arrangement is observed at synapsis since it would place the
two active sites too far apart to participate in target capture and
strand transfer at points that are only 9 base pairs apart as
discussed below. Furthermore, the disposition of helix 1
in the inhibitor protein would be inconsistent with dimerizationin
this way, because 1 is located between 2
and 7 and would block
the interaction between two molecules across this interface. This
analysis is complicated by the fact that a synaptic complex of
Tn5 transposase is likely to be dimeric, whereas a synapticcomplex
of HIV-1 integrase is likely to be tetrameric. Due tothese different stoichiometries,
the protein-protein interactionsin integrase and Tn5 transposase
synapses may be completelydissimilar.
The recent observation that the strand cleavage reaction proceeds through
a hairpin intermediate also places constraints on the arrangement
of the catalytic domains at synapsis and strand transfer (27).3
The presence of a hairpin intermediate explains how a single active
site can cut two strands of DNA. The initial cleavage presumably occurs
via attack of a water molecule, on the first strand of DNA to
leave a 3' OH group. Thereafter the resultant 3' OH attacks the
complementary strand to form a hairpin that is subsequently cleaved
by the attack of a second water molecule to expose the 3' OH
group. It seems seems likely that each of these phosphoryl transfer
reactions utilizes, in whole or in part, the same constellation of
metal ions and protein ligands in an enzymatically similar manner.
In all probability, the same active site components are responsible
for activation of the 3' OH group in the strand transfer reaction
as in the cleavage reaction and implies that the nucleophilic 3'
OH group will be bound close to the base of the canyon that is
proposed to enclose the OE DNA. Since the strand transfer reactionsoccur
at sites 9 base pairs apart on opposite strands, this impliesthat the synaptic
complex delivers the two attacking 3' OH groupson approximately opposite
sides of the DNA helix, depending onthe structure of the intervening bases.
Concerted strand cleavagerequires that both 3' OH groups approach the target
DNA at thesame time. Interestingly, the arrangement of the active site
canyonsobserved in the inhibitor dimer complex precludes such an attacksince
they lie approximately perpendicular to the 2-fold axisof the dimer (Fig.
3b).
This disposition of the active sites wouldnot be able to deliver the 3'
OH groups to an undistorted sectionof target DNA because the OE DNA would
block access to the targetDNA. It is predicted that the catalytic core
of the transposasemust be reoriented relative to that observed in the inhibitordimer
complex to allow direct approach of the active sites tothe target DNA.
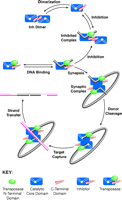
A schematic drawing describing some of these ideasis shown in Fig.
6.
View larger version (25K):
[in
this window]
[in
a new window] |
Fig. 6.
Schematic model for the mechanisms of inhibition and transposition in
the Tn5 system. Transposase is depicted as a three-domain protein
consisting of an N-terminal DNA binding domain (green), the catalytic
core domain (blue, with a yellow dot showing the relative
position of the active site and a groove indicating where DNA binding might
occur), and the C-terminal domain (red). The inhibitor protein lacks
the N-terminal domain of transposase. The
top half of the figure
suggests a mechanism for inhibition. The interaction between the C-terminal
domains of the inhibitor protein (top left) is preserved in the
inhibited complex containing one molecule of inhibitor protein and one
molecule of DNA-bound transposase (top right). The bottom half
of the figure suggests a mechanism for transposition. Starting from a complex
of one molecule of transposase bound to each end of the transposable element,
the synaptic complex is suggested to form by dimerization (lower right).
This representation is not meant to imply the precise relationship of the
transposase subunits in the complex, other than to suggest that the dimer
interface in the synaptic complex is different than that observed in the
inhibited complex. The remainder of the figure is consistent with earlier
models for transposition (5). |
|
Evidence that interactions between the catalytic domain and the C-terminal
dimerization domain are important for transposition is provided
by the phenotype of mutations associated with helix7,
Glu350-Ala378, which forms the connection between
these two domains. Mutation of Leu372 to proline
in the transposase results in a hypertransposing phenotype that
is highly trans-active (65). The mutation maps
to a region, amino acids 369-387, that was postulated to be
important for positioning or stabilization of a dimerization
domain (38, 65). Since Leu372
is located in the middle of the helix adjacent to another prolineresidue,
it is anticipated that introduction of a proline residueat this point will
either cause a greater distortion of the helixor alter the relationship
between the catalytic and C-terminaldomains.
Conclusions-- The structure of transposase Tn5 inhibitor
protein described here answers many of the obvious questions concerning
its tertiary structure and the location and disposition of the
catalytic residues. There remain, however, many unanswered questions
concerning the relationship of this structure to the biological
function of the transposase. It is clear that the conformation
of the catalytic domain and its relationship to the dimerization
interface must be different in the synaptic complex relative
to that seen in the Tn5 inhibitor protein since in the
latter the active sites are too far apart. It seems highly likely
that the interaction of the transposase with the OE DNA increases
the binding affinity of the protein toward a second transposase-OE
DNA complex and that this interaction induces concerted excision
of the transposon. The present structure limits the possibilities
for how this can be accomplished. As such the current study
provides a stepping stone toward understanding the molecular
basis of transposition by the Tn5 transposase.
 §,
§, ,
,