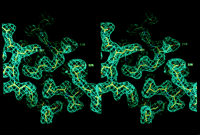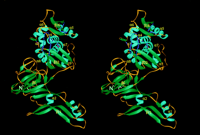Xiaoqun Duan1, Frederick
S. Gimble3, and Florante A. Quiocho1,2
1 Structural and Computational Biology, Molecular
Biophysics Program
2 Howard Hughes Medical Institute, Department
of Biochemistry, Baylor College of Medicine, Houston, Texas 77030
3 Center for Macromolecular Design, Institute
of Biosciences and Technology, and Department of Biochemistry and Biophysics,
Texas A & M University, Houston, Texas 77030
Corresponding author: Florante
A. Quiocho, 713 798 6565 (phone), 713 798 8516 (fax), faq@dino.bcm.tmc.edu.
| Summary |
|---|
| Summary | Introduction | Results and Discussion | Procedures | References |
PI-SceI is a bifunctional yeast protein that propagates its mobile
gene by catalyzing protein splicing and site-specific DNA double-strand
cleavage. Here, we report the 2.4 Å crystal structure
of the PI-SceI protein. The structure is composed of two separate
domains (I and II) with novel folds and different functions.
Domain I, which is elongated and formed largely from seven ß
sheets, harbors the N and C termini residues and two His residues
that are implicated in protein splicing. Domain II, which is
compact and is primarily composed of two similar ![]() /ß
motifs related by local two-fold symmetry, contains the putative
nuclease active site with a cluster of two acidic residues and
one basic residue commonly found in restriction endonucleases.
This report presents prototypic structures of domains with single
endonuclease and protein splicing active sites.
/ß
motifs related by local two-fold symmetry, contains the putative
nuclease active site with a cluster of two acidic residues and
one basic residue commonly found in restriction endonucleases.
This report presents prototypic structures of domains with single
endonuclease and protein splicing active sites.
| Introduction |
|---|
| Summary | Introduction | Results and Discussion | Procedures | References |
PI-SceI is a 454 amino acid (Mr 50 k) bifunctional protein
encoded by a mobile selfish DNA at the VMA1 locus of
S. cerevisiae (![]()
![]()
![]()
![]()
![]()
![]()
Based on conserved sequence motifs or blocks, PI-SceI is related
to two different groups of proteins found in organisms from
each phylogenetic kingdom (for a review, see ![]()
![]()
![]()
Protein splicing of the yeast VMA1 intein and others requires
catalytic elements present in the intein and the first residue
of the C-terminal extein (![]()
![]()
![]()
![]()
To initiate intein homing, PI-Sce I cuts the yeast genome exclusively
at the VMA1 locus (![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() 60°-75°)
when bound to the entire recognition site (
60°-75°)
when bound to the entire recognition site (![]()
![]()
Here, we report the 2.4 Å X-ray crystal structure of the PI-SceI protein. The data clearly reveal the structural and functional duality of the enzyme. Amino acid residues comprising the nucleolytic active site, identified by a cluster of charged residues that are conserved in the protein family, reside in one domain, while those that participate in protein splicing are located in the other. Preliminary docking of a DNA model revealed new features of molecular recognition. Furthermore, examination of the structure immediately suggests an evolutionary model that explains the association of the two disparate activities.
| Results and Discussion |
|---|
| Summary | Introduction | Results and Discussion | Procedures | References |
Structure and Novel Domain Motifs
The three-dimensional structure of PI-SceI, the first for a homing
endonuclease and a protein generated by protein splicing, was
determined by multiwavelength anomalous dispersion (MAD) and
has been refined at 2.4 Å resolution to an R factor and
an Rfree of 19.2% and 23.8%, respectively (Table
1). A portion of the electron density is shown in Figure
1. The two independent molecules within the asymmetric unit
are very similar and can be overlapped with an rms deviation
of about 1 Å between ![]() carbons. The structure is composed of two separate domains (I
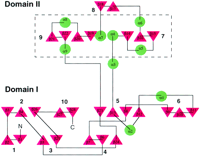
and II) connected by two peptide segments (Figure 2 and
Figure 3). Both domains possess not only unusual folds
but also different functional sites located on the opposite
sides of the molecule. Domain I, comprising the first 182 and
the last 44 residues, is an unusual elongated domain (about
3:1 axial ratio) composed almost entirely of ß sheets
(19 strands). The intervening continuous segment comprising
residues 183-410 adopts an almost equal mixture of helices (7)
and strands (9) forming the compact and globular domain II.
A search (
carbons. The structure is composed of two separate domains (I
and II) connected by two peptide segments (Figure 2 and
Figure 3). Both domains possess not only unusual folds
but also different functional sites located on the opposite
sides of the molecule. Domain I, comprising the first 182 and
the last 44 residues, is an unusual elongated domain (about
3:1 axial ratio) composed almost entirely of ß sheets
(19 strands). The intervening continuous segment comprising
residues 183-410 adopts an almost equal mixture of helices (7)
and strands (9) forming the compact and globular domain II.
A search (![]()
|
||||
|
||||
|
||||
| Table 1. Summary of Crystallographic
Analysis
View this table: |
The 19 strands in domain I are incorporated into a total of 7
closely packed ß sheets approximately divided into two dissimilar
underlying substructures (Figure 2 and Figure
3). The elongated structure of domain I is primarily due
to side-by-side arrangement of sheets 2, 10, 5, and 6, which
constitute one substructure, with sheet 6 providing a long extension.
The other shorter substructure, comprised of sheets 1, 3, and
4, form a ß sandwich with sheets 2, 10, and 5 (Figure
2). As domain I harbors both terminal residues (Cys-1 and
Asn-454) (Figure 2 and Figure
4A), which are conserved between various inteins and are
essential for protein splicing (![]()
|
||||
The compact domain II is mainly built up from two sub- structures,
each with very similar secondary structure motifs (![]() 4-ß14
4-ß14![]() 5ß15ß16
5ß15ß16![]() 6
and
6
and ![]() 7ß19ß20
7ß19ß20![]() 8ß21ß22
8ß21ß22![]() 9)
(Figure 2 and Figure 3). Moreover,
the two motifs are related by local two-fold symmetry about
an axis between the vertical parallel
9)
(Figure 2 and Figure 3). Moreover,
the two motifs are related by local two-fold symmetry about
an axis between the vertical parallel ![]() 4
and
4
and ![]() 7 with
a relative twist of about 35° (Figure 2). Helices
4 and 7 contain the two dodecapeptide sequences that are distinguishing
characteristics of homing endonucleases and maturases (
7 with
a relative twist of about 35° (Figure 2). Helices
4 and 7 contain the two dodecapeptide sequences that are distinguishing
characteristics of homing endonucleases and maturases (![]()
![]()
![]()
![]() 5
and
5
and ![]() 6 in one motif
related to
6 in one motif
related to ![]() 8 and
8 and ![]() 9,
respectively, in the other). The approximate symmetry is also
clearly apparent between the two ß sheets in both motifs
that flank the C-terminal ends of the two parallel helices and
that form a concave twisted canopy above the symmetry-related helices
(Figure 2). (Although the segment following
9,
respectively, in the other). The approximate symmetry is also
clearly apparent between the two ß sheets in both motifs
that flank the C-terminal ends of the two parallel helices and
that form a concave twisted canopy above the symmetry-related helices
(Figure 2). (Although the segment following ![]() 4
in one sheet is not quite a sheet strand, it is topologically
equivalent to ß14 of the other sheet [ Figure
2 and Figure 3]). The similarity of the
two motifs is evident from superpositioning of the
4
in one sheet is not quite a sheet strand, it is topologically
equivalent to ß14 of the other sheet [ Figure
2 and Figure 3]). The similarity of the
two motifs is evident from superpositioning of the ![]() carbon atoms of the 63 pairs of residues in the overlapped secondary
structures of both motifs, which show an rms deviation of 1.7
Å. The resulting overlapped sequences reveal 22% identity
and 46% similarity.
carbon atoms of the 63 pairs of residues in the overlapped secondary
structures of both motifs, which show an rms deviation of 1.7
Å. The resulting overlapped sequences reveal 22% identity
and 46% similarity.
Endonuclease Active Site
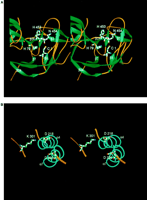
The nuclease active site apparently resides in domain II, at the
C-terminal ends of the parallel helices 4 and 7. This location is
consistent with several pieces of evidence. Two Asp residues (Asp-218
and Asp-326) that are located at the C termini of the parallel
helices and Lys-301, found in a loop immediately after ß18,
form a charged cluster (Figure 2 and Figure
4B) that bears similarity with those commonly seen in the
catalytic sites of homodimeric restriction endonucleases with
previously determined three-dimensional structures (EcoRI, PvuII,
EcoRV, and BamHI; for a review, see ![]()
![]() carbons.
The Asp-218 and Asp-326 residues in PI-SceI occur at positions
within the dodecapeptide motifs that are extremely conserved
as acidic residues among the related homing endonucleases and
maturases (
carbons.
The Asp-218 and Asp-326 residues in PI-SceI occur at positions
within the dodecapeptide motifs that are extremely conserved
as acidic residues among the related homing endonucleases and
maturases (![]()
![]()
![]() helices that
correctly position the active site residues. Lys-301 is a conserved
residue within a separate motif found in inteins (Block D, Pietrokovski,
1994) and several maturases. Finally, substitutions of the two
Asp residues by site-directed mutagenesis abolish DNA cleavage
but not binding (
helices that
correctly position the active site residues. Lys-301 is a conserved
residue within a separate motif found in inteins (Block D, Pietrokovski,
1994) and several maturases. Finally, substitutions of the two
Asp residues by site-directed mutagenesis abolish DNA cleavage
but not binding (![]()
In spite of profound differences in the overall structures of the
restriction endonucleases and PI-SceI (discussed below), the
similar active site arrangement suggests an analogous hydrolytic mechanism.
Asp-218 and Asp-326 of PI-SceI appear to be structurally equivalent
to acidic residues in EcoRI, EcoRV, BamHI, and PvuII (![]()
![]()
![]()
![]()
Because PI-SceI is a monomer in solution (![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Potential DNA-Binding Sites and DNA Docking
The nature of the areas around the putative active site in domain
II indicates potential sites for DNA binding. The obvious areas
include the exposed surfaces of the two symmetry-related ß
sheets flanking the two Asp active site residues and the ß-hairpin
loops (between ß15 and ß16 and between ß21
and ß22) above the sheets (Figure 2). Loops
are very often seen in structures of DNA-binding proteins involved
in interacting with either the major or minor groove of DNA,
including those of restriction endonucleases (![]()
![]()
![]()
![]()
![]()
![]() 4)
and ß14 and with the loop preceeding ß16 that is
above the sheet (Figure 2 and Figure 3).
The two sites after the second dodecapeptide repeat align with
the loop between ß19 and ß20 and with the large
loop between ß21 and ß22 above the sheet (Figure
2 and Figure 3). Furthermore, mutations
located within the large loops and ß sheets interfere
with substrate binding (F. S. G., unpublished data).
4)
and ß14 and with the loop preceeding ß16 that is
above the sheet (Figure 2 and Figure 3).
The two sites after the second dodecapeptide repeat align with
the loop between ß19 and ß20 and with the large
loop between ß21 and ß22 above the sheet (Figure
2 and Figure 3). Furthermore, mutations
located within the large loops and ß sheets interfere
with substrate binding (F. S. G., unpublished data).
To help comprehend the interaction of PI-SceI with its lengthy
recognition site (31 bp or longer; ![]()
![]() ),
we carried out a preliminary docking of a DNA onto the structure
using the program GRASP (
),
we carried out a preliminary docking of a DNA onto the structure
using the program GRASP (![]()
![]() )
and right (or plus) sequences, respectively, of the center of
the cleavage site. This docking arrangement of the DNA takes
into account the experimental observation that the left sequence
requires fewer base pairs than the right for DNA binding and
cleavage (
)
and right (or plus) sequences, respectively, of the center of
the cleavage site. This docking arrangement of the DNA takes
into account the experimental observation that the left sequence
requires fewer base pairs than the right for DNA binding and
cleavage (![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
|
||||
In docking the DNA, it became apparent that the size or diameter
of globular domain II is insufficient to accommodate the entire
30 bp DNA model, even in the presence of the bend. DNA binding
would require the participation of domain I. The docking model
indicates that domain II can recognize about 14 bp (from about
-8 bp to +6 bp of the cleavage site). The additional 16 or more
bp on the plus, or right, sequence of the cleavage site extends
to the arm of domain I, which contains a high concentration
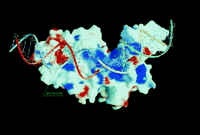
of clusters of intense positive charge (Figure 5).
The limited base sequence potentially recognized by domain II
is consistent with the observation that endonucleases related
only to this domain recognize much shorter DNA substrates (![]() 14-20
bp). In addition, the sharp bend in the DNA, which has been
experimentally observed in complexes of PI-SceI with DNA, may
very well be due to the presence of the elongated domain I which,
as the structure indicates, adopts a roughly equivalent bend
relative to domain II.
14-20
bp). In addition, the sharp bend in the DNA, which has been
experimentally observed in complexes of PI-SceI with DNA, may
very well be due to the presence of the elongated domain I which,
as the structure indicates, adopts a roughly equivalent bend
relative to domain II.
The Protein Splicing Catalytic Site
The structure of the PI-SceI intein represents the excised end
product of protein splicing. The positions of the key junction
amino acid residues (Figure 4A) identified by mutation
are entirely consistent with their proposed roles in the reaction
pathway of self-splicing (![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Evolutionary Implications of the Structure
The bipartite domain structure of PI-SceI is likely paralleled
by a separation of the protein splicing and endonucleolytic
cleavage activities. In the case of the related PI-TliI intein,
it has been demonstrated that mutations that abolish one activity
have little or no effect on the other (![]()
![]()
Based on these observations, we hypothesize that the VMA1 intein
is encoded by a composite gene that resulted from the invasion
of an endonuclease ORF into a preexisting gene that encoded
a protein with protein splicing activity or that later evolved
this activity. The endonuclease ORF is likely to have been the
mobilizing entity rather than the protein splicing ORF, because
endonucleolytic activity is required for intein and intron homing.
Furthermore, the fact that the endonuclease ORF is embedded
in the middle of the protein splicing ORF provides additional
circumstantial evidence that it was the invading entity. Once
these genes were fused, we speculate that the entire endonuclease-splicing
ORF functioned as a mobile element that inserted into the VMA1
locus. The symbiotic association of the endonuclease ORF with
the splicing ORF benefits both entities. The endonuclease ORF
is associated with a gene that encodes a polypeptide that safely
removes itself and the endonuclease from the vacuolar H+-ATPase
host protein and prevents any deleterious effects to the host.
By allying itself with an endonuclease ORF, the splicing gene
is assured of mobility within the same species and eventually
to new species as well, perhaps by horizontal transmission.
This scenario is analogous to proposed models that explain the
association of Group I introns and endonuclease ORFs (![]()
![]()
![]()
Comparison of Endonuclease Structures
With the exception of the similarity with the catalytic charged
residue triad, PI-SceI endonuclease is very different from EcoRI,
PuvII, EcoRV, and BamHI restriction endonucleases, which together
do not show common features. The Type II restriction enzymes
function as homodimers, with each subunit interacting with a
separate half-site of a palindromic sequence, whereas PI-SceI
is a monomeric protein that contacts an extended asymmetric
site. This points to a difference in strategy; the use of a
palindromic target limits the size of the recognition site and
the selectivity, but the dimeric composition of restriction
enzymes permits a smaller subunit size (![]()
![]()
The three-dimensional structure reported here paves the way for investigations that will elucidate the molecular recognition functions and catalytic activities of PI-SceI. Further experiments (e.g., site-directed mutagenesis) suggested by the structure will enable us to test several predictions that result from the detail that it provides.
| Experimental Procedures |
|---|
| Summary | Introduction | Results and Discussion | Procedures | References |
Protein Purifications and Crystallization
The procedures for overexpression and purification of recombinant
wild-type PI-SceI have been previously described (![]()
![]()
![]()
![]()
![]()
Both wild-type and Se-Met PI-SceI were crystallized at room temperature using the hanging drop vapor diffusion method over a reservoir solution containing 4% PEG 6K, 10 mM ß-mercaptoethanol (ßME), 3 mM CdCl2, 1 mM MgCl2, and 100 mM Tris (pH 8.5). The 2 µl drops of the protein (8 mg/ml in 5 mM ßME and 20 mM Tris [pH 8.0]) were mixed with an equal volume of the reservoir solution. Relative to the wild-type protein, Se-Met PI-SceI crystals were bigger and easier to reproduce and showed diffraction to higher resolution. The crystals belong to space group P21 with unit cell parameters a = 59.6 Å, b = 102.9 Å, c = 87.4 Å, and ß = 94.3° for the wild-type protein crystal measured with laboratory area detector, and a = 59.8 Å, b = 102.4 Å, c = 87.1 Å, and ß = 94.1° for the Se-Met protein measured using synchrotron data. There are two molecules per asymmetric unit, and the solvent content is about 53%. Crystals for data collection were stabilized in the mother liquor, which contained 20% glycerol, and flashcooled to -170°C in liquid nitrogen.
Data Collection
MAD data were collected from one frozen crystal at the HHMI X4A
beam line of the National Synchrotron Light Source (NSLS). The
optimal wavelengths for Se data collection at NSLS were determined
by measuring fluorescence scan with a scintillation counter
using the frozen PI-SceI crystal from which data were taken.
The oscillation data were then collected at the absorption edge
(0.9794 Å), peak (0.9790 Å), and remote peak (0.9656
Å) using inverse beam geometry with an oscillation angle
of 1.2°. The data were indexed and integrated using the
program DENZO and scaled within each wavelength using SCALEPACK (![]()
Structure Determination
MAD phasing using reflections from 10 to 2.6 Å was done
using the MADSYS suite of programs (![]()
As there are 8 Met residues in the wild-type PI-SceI, we expect
a total of 16 selenium sites in the two Se-Met PI-SceI molecules
within the asymmeteric unit. The positions of 8 initial selenium
atoms were determined from the anomalous difference Patterson
map of the peak wavelength with SHELXS (![]()
The 14 total selenium sites were refined by ASLSQ and input into
MADFAZ to obtain phases at 2.6 Å resolution (Table
1). The resulting 2.6 Å electron density map showed
clear solvent boundary and several secondary structures and
confirmed the presence of two molecules in the asymmetric unit.
Density modification and two-fold noncrystallographic averaging
using SOLV and AVER options, respectively, of "dm"
(![]()
Model Building and Refinement
The skeleton generated by BONE (![]()
![]()
![]()
![]()
![]()
![]() 6,
and 369-374 between ß21 and ß22 (Figure 2
and Figure 3). The coordinates have been deposited
in the Protein Data Bank (ID code 1VDE).
6,
and 369-374 between ß21 and ß22 (Figure 2
and Figure 3). The coordinates have been deposited
in the Protein Data Bank (ID code 1VDE).
Structure Analysis
The correctness of the final model was verified by examining its
stereochemistry using the program PROCHECK (![]()
![]()
![]()
![]()
| Acknowledgments |
|---|
Correspondence regarding this paper should be addressed to F. A. Q. We thank C. Ogata of HHMI/Brookhaven NSLS and A. Nickitenko, A. Hodel, and Z. Wang of the F. A. Q. lab for assistance with data collection at the HHMI X4A beam line of NSLS and for helpful discussions; W. Hendrickson and A. DiGabriele for providing strain DL41 (DE3) and protocols; W. Meador for X-ray technical assistance; and J. Wang and E. Golunski for assistance in protein purification. This work was supported by National Institutes of Health grant R29 GM50815 (F. S. G.), funds from the Institute of Biosciences and Technology (F. S. G.), and funds from the Offices of Research and Information Technology of Baylor College of Medicine (F. A. Q.). X. D. is supported by an NIH-NIGMS Pre-Doctoral Training Grant (GM08280) to the Houston Area Molecular Biophysics Program. F. A. Q. is an Investigator of the Howard Hughes Medical Institute.
Received March 12, 1997; revised April 8, 1997.
| References |
|---|
| Summary | Introduction | Results and Discussion | Procedures | References |
Aggarwal, A.K. (1995). Structure and function of restriction endonucleases. Curr. Opin. Struct. Biol. 5, 11-19.[Medline]
Baldwin, G.S., Vipond, I.B., and Halford, S.E. (1995). Rapid reaction analysis of the catalytic cycle of the EcoRV restriction endonuclease. Biochemistry 34, 705-714.[Medline]
Belfort, M. (1989). Bacteriophage introns: parasites within parasites? Trends Genet. 5, 209-213.[Medline]
Bremer, M.C.D., Gimble, F.S., Thorner, J., and Smith, C.L. (1992). VDE endonuclease cleaves Saccharomyces cerevisiae genomic DNA at a single site: physical mapping of the VMA1 gene. Nucl. Acids Res. 20, 5484.
Brünger, A.T. (1992). X-PLOR Version 3.1: A System for X-Ray Crystallography and NMR., New Haven, CT: Yale University Press.
Carson, M. (1991). Ribbons 2.0. J. Appl. Cryst. 24, 958-961.
Cheng, X., Balendiran, K., Schildkraut, I., and Anderson, J.E. (1994). Structure of PvuII endonuclease with cognate DNA. EMBO J. 13, 3927-3935.[Medline]
Chong, S., Shao, Y., Paulus, H., Benner, J., Perler, F.B., and Xu, M.-Q. (1996). Protein splicing involving the Saccharomyces cerevisiae VMA intein. J. Biol. Chem. 271, 22159-22168.[Medline]
Cooper, A.A., Chen, Y.-J., Lindorfer, M.A., and Stevens, T.H. (1993). Protein splicing of the yeast TFP1 intervening protein sequence: a model of self-excision. EMBO J. 12, 2575-2583.[Medline]
Cooper, A.A., and Stevens, T.H. (1995). Protein splicing: self-splicing of genetically mobile elements at the protein level. Trends Biochem. Sci. 20, 351-356.[Medline]
Cowton, K. (1994). `dm': an automated procedure for phase improvements by density modifications. Joint CCP4 and ESF-EACBM Newslett. Protein Cryst. 31, 34-38.
Gimble, F.S., and Stephens, B.W. (1995). Substitutions in conserved dodecapeptide motifs that uncouple the DNA binding cleavage activities of PI-SceI endonuclease. J. Biol. Chem. 270, 5849-5856.[Medline]
Gimble, F.S., and Thorner, J. (1992). Homing of a DNA endonuclease gene by meiotic gene conversion in Saccharomyces cerevisiae. Nature 357, 301-306.[Medline]
Gimble, F.S., and Thorner, J. (1993). Purification and characterization of VDE, a site-specific endonuclease from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 268, 21844-21853.[Medline]
Gimble, F.S., and Wang, J. (1996). Substrate recognition and induced DNA distortion by the PI-SceI endonuclease, an enzyme generated by protein splicing. J. Mol. Biol. 263, 163-180.[Medline]
Hendrickson, W.A., Horton, J.R., and LeMaster, D.M. (1990). Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD); a vehicle for direct determination of three-dimensional structure. EMBO J. 9, 1665-1572.[Medline]
Hendrickson, W.A. (1991). Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science 254, 51-58.[Medline]
Henke, R.M., Butow, R.A., and Perlman, P.S. (1995). Maturase and endonuclease functions depend on separate conserved domains of the bifunctional protein encoded by the group I intron aI4a of yeast mitochondrial DNA. EMBO J. 14, 5094-5099.[Medline]
Hensgens, L.A.M., Bonen, L., de Haan, M., van der Horst, G., and Grivell, L. (1983). Two intron sequences in yeast mitochondrial COX1 gene: homology among URF-containing introns and strain-dependent variation in flanking exons. Cell 32, 379-389.[Summary]
Hirata, R., Ohsumi, Y., Nakano, A., Kawasaki, H., Suzuki, K., and Anraku, Y. (1990). Molecular structure of a gene, VMA1, encoding the catalytic subunit of H+-translocating adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J. Biol. Chem. 265, 6726-6733.[Medline]
Hirata, R., and Anraku, Y. (1992). Mutations at the putative junction sites of the yeast VMA1 protein, the catalytic subunit of the vacuolar membrane H+-ATPase, inhibit its processing by protein splicing. Biochem. Biophys. Res. Comm. 188, 40-47.[Medline]
Hodges, R.A., Perler, F.B., Noren, C.J., and Jack, W.E. (1992). Protein splicing removes intervening sequences in an archaea DNA polymerase. Nucl. Acids Res. 20, 6153-6157.
Holm, L., and Sander, C. (1995). Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 20, 478-480.[Medline]
Jones, T.A., Zou, J.Y., Kjelgaard, M., and Cowan, S.W. (1991). Improved methods for building protein models in electron-density maps and the location of errors in these models. Acta Cryst. A 47, 110-119.
Kane, P.M., Yamashiro, C.T., Wolczyk, D.F., Neff, N., Goebl, M., and Stevens, T.H. (1990). Protein splicing converts the yeast TFP1 gene product to the 69-kD subunit of the vacuolar H+-adenosine triphosphatase. Science 250, 651-657.[Medline]
Kabsch, W., and Sander, C. (1983). Dictionary of protein secondary structure: pattern of hydrogen-bonded and geometrical features. Biopolymers 22, 2577-2637.[Medline]
Kostrewa, D., and Winkler, F.K. (1995). Mg2+ binding to the active site of EcoRV endonuclease: a crystallographic study of complexes with substrate and product DNA at 2 Å resolution. Biochemistry 34, 683-696.[Medline]
Lambowitz, A.M. (1989). Infectious introns. Cell 56, 323-326.[Summary]
Lawskowski, R.A., McArthur, M.W., Moss, D.S., and Thorton, J.M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 282-291.
Loizos, N., Tillier, E.R.M., and Belfort, M. (1994). Evolution of mobile group I introns: recognition of intron sequences by an intron- encoded endonuclease. Proc. Natl. Acad. Sci. USA 91, 11983-11987.[Medline]
Luthy, R., Bowie, J.U., and Eisenberg, D. (1992). Assessment of protein models with three-dimensional profiles. Nature 356, 83-85.[Medline]
Lykke-Andersen, J., Garrett, R.A., and Kjems, J. (1996). Protein footprinting approach to mapping DNA binding sites of two archaeal homing enzymes: evidence for a two-domain protein structure. Nucl. Acids Res. 24, 3982-3989.
Michel, F., Jacquier, A., and Dujon, B. (1982). Comparison of fungal mitochondrial introns reveals extensive homologies in RNA secondary structure. Biochimie 64, 867-881.[Medline]
Mueller, J.E., Bryk, M., Loizos, N., and Belfort, M. (1993). Homing endonucleases. In Nucleases, S.M. Linn, R.S. Lloyd, and R.J. Roberts, eds. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), pp. 111-143.
Nicholls, A., Bharadwaj, R., and Honig, B. (1993). GRASP: graphical representation and analysis of surface properties. Biophys. J. 64, 166-170.
Otwinowski, Z., and Minor, W. (1997). Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzymol. 276, 307-326.
Perler, F.B., Olsen, G.J., and Adam, E. (1997). Compilation and analysis of intein sequences. Nucl. Acids Res. 25, 1087-1094.
Pietrokovski, S. (1994). Conserved sequence features of inteins (protein introns) and their use in identifying new inteins and related proteins. Protein Sci. 3, 2340-2350.[Medline]
Raumann, B.E., Rould, M.A., Pabo, C.O., and Sauer, R.T. (1994). DNA recognition by ß-sheets in the Arc repressor-operator crystal structure. Nature 367, 754-757.[Medline]
Read, R.J. (1986). Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Cryst. A 47, 110-119.
Sack, J.S. (1988). CHAIN![]() a
crystallography molecular graphics program. J. Mol. Graphics 6, 224-245.
a
crystallography molecular graphics program. J. Mol. Graphics 6, 224-245.
Schleif, R. (1988). DNA binding by proteins. Science 241, 1182-1187.[Medline]
Sheldrick, G.M. (1991). Heavy atom location using SHELXS-90. In Isomorphous Replacement and Anomalous Scattering: Proceedings of the CCP4 Study Weekend, 25-26 January 1992, W. Wolf, P. Evans, and A.G.W. Leslie, eds. (Warrington, UK: SERC Daresbury Laboratory).
Skowron, P., Kaczorowski, T., Tucholski, J., and Podhajska, A.J. (1993). Atypical DNA-binding properties of class-IIS restriction endonucleases: evidence for recognition of the cognate sequence by a FokI monomer. Gene 125, 1-10.[Medline]
Somers, W.S., and Phillips, S.E.V. (1992). Crystal structure of the met repressor-operator complex at 2.8 Å resolution reveals DNA recognition by ß-strands. Nature 367, 754-757.
Vipond, I.B., Baldwin, G.S., and Halford, S.E. (1995). Divalent metal ions at the active sites of the EcoRV and EcoRI restriction endonucleases. Biochemistry 34, 697-704.[Medline]
Waring, R.B., Davies, R.W., Scazzocchio, C., and Brown, T.A. (1982). Internal structure of a mitochondrial intron of Aspergillus nidulans. Proc. Natl. Acad. Sci. USA 79, 6332-6336.[Medline]
Waugh, D.S., and Sauer, R.T. (1993). Single amino acid substitutions uncouple the DNA binding and strand scission activities of FokI endonuclease. Proc. Natl. Acad. Sci. USA 90, 9596-9600.[Medline]
Wende, W., Grindl, W., Christ, F., Pingoud, A., and Pingoud, V. (1996). Binding, bending and cleavage of DNA substrates by the homing endonuclease PI-SceI. Nucl. Acids Res. 24, 4123-4132.