Kjell Håkansson1,
Aidan J. Doherty1, Stewart Shuman2,
and Dale B. Wigley1
1 Laboratory of Molecular Biophysics, University
of Oxford, Oxford, OX1 3QU, United Kingdom
2 Molecular Biology Program, Memorial Sloan-Kettering
Institute, New York, New York 10021
Corresponding author: Dale
B. Wigley, 01865 275381 (phone), 01865 275182 (fax), wigley@biop.ox.ac.uk.
| Summary |
|---|
| Summary | Introduction | Results and Discussion | Procedures | References |
We have solved the crystal structure of an mRNA capping enzyme at 2.5 Å resolution. The enzyme comprises two domains with a deep, but narrow, cleft between them. The two molecules in the crystallographic asymmetric unit adopt very different conformations; both contain a bound GTP, but one protein molecule is in an open conformation while the other is in a closed conformation. Only in the closed conformation is the enzyme able to bind manganese ions and undergo catalysis within the crystals to yield the covalent guanylated enzyme intermediate. These structures provide direct evidence for a mechanism that involves a significant conformational change in the enzyme during catalysis.
| Introduction |
|---|
| Summary | Introduction | Results and Discussion | Procedures | References |
Eukaryotic mRNA is capped at the 5' end, by a 7-methyl-GMP moiety
(see Shuman, 1995 for a review). This cap plays an important
role in RNA synthesis and function and may also help to protect
the mRNA from nucleolytic digestion. Since the capping of mRNA
has been shown to be an essential activity in yeast (![]()
![]()
The capping reaction involves three separate processes that are
catalyzed by three different enzyme activities. The first step
involves the removal of the 5'-ter-minal phosphate from the
triphosphate tail by an RNA triphosphatase. The reaction is
usually followed by guanylation and then methylation, although the
order of these reactions can apparently be reversed in some viruses
(![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Although the sequence of reactions that occurs during catalysis
by capping enzymes is known, much less is known about the molecular
details of these processes. Mutational analysis of capping enzymes
and ligases has indicated likely roles for some residues (![]()
![]()
![]()
![]()
![]()
![]()
![]()
| Results and Discussion |
|---|
| Summary | Introduction | Results and Discussion | Procedures | References |
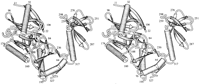
Structure of the Protein
Details of the structure determination and refinement are presented
in Table 1. Residues 11-327 of the 330 amino acids
of the protein were defined in our final model. The enzyme comprises
two domains with a cleft between them. Domain 1 consists of
residues 11-236 and 319-327, while domain 2 comprises residues
238-317 (Figure 1). A large part of domain
1 has a topology similar to that of a group of ATP-binding proteins
that includes DNA ligase, although the three-dimensional structure
is rather different. Furthermore, the capping enzyme contains
a 50 residue extension at the N terminus that is not conserved
in ligases, which forms an additional two-stranded ß sheet
and an ![]() helix. Domain
1 contains the GTP-binding site and the active site lysine to
which the GMP becomes attached during catalysis. A series of
conserved motifs, which have been proposed previously to indicate
a conservation of structure across a wide family of nucleotidyltransferases
that includes capping enzymes and ligases (
helix. Domain
1 contains the GTP-binding site and the active site lysine to
which the GMP becomes attached during catalysis. A series of
conserved motifs, which have been proposed previously to indicate
a conservation of structure across a wide family of nucleotidyltransferases
that includes capping enzymes and ligases (![]()
![]()
![]() helix. The last few residues of the protein (319-327) return
across the cleft between the domains to form an amphipathic
helix. The last few residues of the protein (319-327) return
across the cleft between the domains to form an amphipathic
![]() helix that packs against
domain 1.
helix that packs against
domain 1.
|
||||
| Table 1. Summary of Crystallographic
Structure Analysis
View this table: |
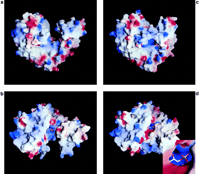
The crystal structure of DNA ligase revealed that the enzyme comprised
two domains with a cleft between them. Calculations of the electrostatic
surface potential (![]()
![]()
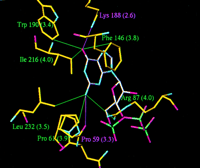
The positions of the base and the ribose of the bound GTP are similar to those of ATP bound at the active site of ligase and involve many similar contacts with residues from the conserved motifs. However, the specificity of the capping enzyme for GTP over ATP seems to be determined by residues outside of these motifs (Figure 2). The 6-oxo group of the guanine ring is involved in hydrogen bonds with the side chain of Lys-188 and van der Waals interactions with the side chain of Trp-190, while the 2-amino group is involved in a rather long hydrogen bond to the main chain carbonyl of Pro-59. None of these interactions would be possible with ATP. The specificity of DNA ligase for ATP over GTP was explained by interactions between the 6-amino group of the adenine ring and a glutamate residue (Glu-32), that is conserved in ligase sequences, and with the main chain carbonyl of Ile-33.
|
||||
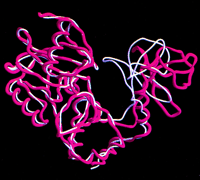
Surprisingly, the two molecules of the capping enzyme in the crystallographic asymmetric unit adopt very different conformations (Figure 3). While one of the enzyme molecules has a deep, but narrow, cleft between the two domains, in the other molecule, this cleft is closed off as a result of a rigid body movement of domain 2. This movement results in a maximum displacement of 13 Å at the tip of the domain. This dramatic difference is important for the catalytic mechanism of the enzyme, and we shall refer to each of these conformations as "open" or "closed."
|
||||
GTP-Binding Sites of the Two Molecules
Although electron density for the bound nucleotide cofactor was
evident at the active sites of both molecules, even the initial
MIR, solvent-flattened electron density maps revealed that they
were different. In light of these differences, we were careful
to exclude these ligands from the refinement until the final
stages, to prevent model bias. The difference electron density
in the final stages of refinement revealed that the active sites
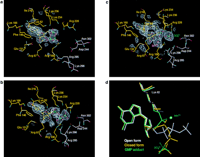
of both enzyme molecules contained GTP (Figure 4).
While the density for the bound GTP was strong for the closed
molecule, it was poorer in the open molecule, with the density
for the ![]() phosphate
being particularly weak. The conformation of the triphosphate
tail of each of the GTP molecules was different (Figure
4). We believe that this observation is important for a
consideration of the enzyme mechanism (see below). Furthermore, the
unexpected discovery of two conformational states of the GTP
complex in the crystal provides direct evidence for a conformational
change in the enzyme during nucleotide binding and catalysis.
phosphate
being particularly weak. The conformation of the triphosphate
tail of each of the GTP molecules was different (Figure
4). We believe that this observation is important for a
consideration of the enzyme mechanism (see below). Furthermore, the
unexpected discovery of two conformational states of the GTP
complex in the crystal provides direct evidence for a conformational
change in the enzyme during nucleotide binding and catalysis.
|
||||
The GTP is bound in the active site of both enzyme molecules, with
the guanosine base in a syn conformation buried in a hydrophobic pocket
between Phe-146 (parallel stacking) and Ile-216. The ribose
hydroxyls make hydrogen bonds with Glu-131 and Arg-87. O6 of
the guanine base is hydrogen bonded to Lys-188 and makes a van
der Waals contact with the edge of the aromatic ring of Trp-190.
The triphosphate chain of the bound GTP makes very few contacts
with the protein in the open form; the ![]() phosphate is hydrogen bonded to the active site lysine Lys-82
and to Lys-234 (Figure 4). Upon domain closure,
the
phosphate is hydrogen bonded to the active site lysine Lys-82
and to Lys-234 (Figure 4). Upon domain closure,
the ![]() phosphate moves
away from Lys-234 and hydrogen bonds to Lys-236. As a result,
the distance between the active site lysine and the
phosphate moves
away from Lys-234 and hydrogen bonds to Lys-236. As a result,
the distance between the active site lysine and the ![]() phosphorus decreases from 4.0 to 3.2 Å. What is perhaps
more important is that the conformation of the entire triphosphate
moiety becomes different from that observed in the open form,
with the ß and
phosphorus decreases from 4.0 to 3.2 Å. What is perhaps
more important is that the conformation of the entire triphosphate
moiety becomes different from that observed in the open form,
with the ß and ![]() phosphates folded away from the active site lysine. The
phosphates folded away from the active site lysine. The ![]() amino group of the lysine and the leaving group are on opposite
sides of the
amino group of the lysine and the leaving group are on opposite
sides of the ![]() phosphorus, which is suitable for in-line attack of the phosphorus
by the lysine. The ß and
phosphorus, which is suitable for in-line attack of the phosphorus
by the lysine. The ß and ![]() phosphates are within hydrogen bonding distance of not only
Arg-106, Arg-228, and Lys-234 of the N-terminal domain, but
also Asp-244, Arg-295, Lys-298, and Asn-302 of the C-terminal
domain. All of the oxygens of the
phosphates are within hydrogen bonding distance of not only
Arg-106, Arg-228, and Lys-234 of the N-terminal domain, but
also Asp-244, Arg-295, Lys-298, and Asn-302 of the C-terminal
domain. All of the oxygens of the ![]() phosphate are bound to positively charged residues in what appears
to be a strong anion-binding site.
phosphate are bound to positively charged residues in what appears
to be a strong anion-binding site.
Soaking of the crystals in harvest solution lacking GTP resulted in rapid diffusion of the GTP from the crystals, such that after two hours, the electron density for GTP could not be observed in either of the two molecules (data not shown). This confirms that the nucleotide moiety was not attached covalently in either molecule.
Formation of the GMP Adduct in the Crystals
The surprising discovery that there were two conformations of
the protein in the crystals led us to investigate the biochemical
reasons for these differences. A close examination of the electron
density around the bound GTP molecules revealed that there was
no candidate for a bound magnesium ion in either protein molecule,
even though 5 mM magnesium chloride was present during the crystallization.
We attribute this to the high salt concentration required for
crystallization. To overcome this effect, we soaked the crystals
in harvest solution containing a high concentration (100 mM)
of manganese chloride for four hours. We chose the more electron
dense manganese rather than magnesium to assist in the identification
of the bound cation in the final difference electron density
maps. Manganese has been shown to be as effective as magnesium
as cofactor for the Chlorella capping enzyme (![]()
After soaking, we examined the electron density of the active sites
of each of the two molecules. The bound GTP in the open molecule
retained the same conformation as observed in the original structure.
Furthermore, there was no evidence of a bound manganese ion
in this site. However, the electron density at the active site
of the closed molecule had undergone significant changes (Figure
4). It was immediately evident that there was a large peak
of difference electron density adjacent to the ![]() phosphate of the nucleotide that we interpret as a bound manganese
ion. In addition, although the electron density for the ß
and
phosphate of the nucleotide that we interpret as a bound manganese
ion. In addition, although the electron density for the ß
and ![]() phosphates
had disappeared, there was now continuous density between the
phosphates
had disappeared, there was now continuous density between the
![]() phosphate and the active
site lysine residue. There were also subtle changes in conformation
of the protein side chains around the active site. All of the
oxygens of the Lys-GMP phosphate are interacting with charged
residues; one with Lys-234 and the other two with Lys-236 and
the manganese ion, respectively. This latter interaction is
the only close contact involving the manganese ion, although
there are other interactions that appear to be mediated by water
molecules. Electron density that we interpret as a sulphate
ion (based upon the environment and the height of the density
peak) is now found in the position occupied previously by the
phosphate and the active
site lysine residue. There were also subtle changes in conformation
of the protein side chains around the active site. All of the
oxygens of the Lys-GMP phosphate are interacting with charged
residues; one with Lys-234 and the other two with Lys-236 and
the manganese ion, respectively. This latter interaction is
the only close contact involving the manganese ion, although
there are other interactions that appear to be mediated by water
molecules. Electron density that we interpret as a sulphate
ion (based upon the environment and the height of the density
peak) is now found in the position occupied previously by the
![]() phosphate. These observations
are consistent with turnover of the enzyme to cleave the bound
GTP and produce the covalent guanylated enzyme intermediate.
Since this change had only taken place in the closed conformation
of the enzyme, we conclude that only this form is catalytically
competent. We presume that this is due to an inability of the
open form of the enzyme to bind the divalent cation that is
required for cleavage of the GTP and to the differences in the
conformation of the triphosphate moiety. The direct consequence
of this observation is that upon binding of GTP, the enzyme
undergoes a conformational change to produce the active form
of the enzyme, which is then able to bind manganese (or magnesium)
ions and promote guanylation.
phosphate. These observations
are consistent with turnover of the enzyme to cleave the bound
GTP and produce the covalent guanylated enzyme intermediate.
Since this change had only taken place in the closed conformation
of the enzyme, we conclude that only this form is catalytically
competent. We presume that this is due to an inability of the
open form of the enzyme to bind the divalent cation that is
required for cleavage of the GTP and to the differences in the
conformation of the triphosphate moiety. The direct consequence
of this observation is that upon binding of GTP, the enzyme
undergoes a conformational change to produce the active form
of the enzyme, which is then able to bind manganese (or magnesium)
ions and promote guanylation.
Mechanistic Implications
Examination of the structures of the two different conformations
of the capping enzyme reveals that while only the open form
seems wide enough to accomodate the RNA substrate, only the
closed form could be guanylated in the presence of excess GTP
and Mn2+ ions. Therefore, since both reactions are catalyzed
by the enzyme, both forms appear to be required in order to
complete GMP transfer from GTP to RNA. The conformational differences
between the ligands in the open and closed forms show how GTP
is positioned optimally for reaction with Lys-82 only in the
closed form of the protein. A similar electrophilic activation
of the ![]() phosphate
of ATP in the active site of tyrosyl tRNA synthetase has been
suggested from model building (
phosphate
of ATP in the active site of tyrosyl tRNA synthetase has been
suggested from model building (![]()
![]() phosphates also
facilitate cleavage of the bond to the leaving group as the
phosphates also
facilitate cleavage of the bond to the leaving group as the
![]() phosphate approaches
the active site lysine. While this conformational activation
requires domain closure and the assistance of domain 2, it is
plausible that a similar phosphate conformation could be achieved
in the analogous capping reaction through interaction between
RNA and enzyme.
phosphate approaches
the active site lysine. While this conformational activation
requires domain closure and the assistance of domain 2, it is
plausible that a similar phosphate conformation could be achieved
in the analogous capping reaction through interaction between
RNA and enzyme.
What other evidence is there for this large conformational change?
One obvious consequence of the domain closure is that a number
of residues in the C-terminal domain are brought into the proximity
of the GTP-binding site. Mutational analysis of an aspartate
residue (Asp-257) in the Saccharomyces cerevisiae capping enzyme
(![]()
![]()
![]()
The structures presented here suggest that the magnesium-binding site can only be formed after domain closure. If a magnesium ion were bound at this site, then conversion of GTP to enzyme-GMP adduct would have taken place. Even though magnesium ions were present in the crystallization medium, the high salt concentration appears to have prevented the binding of magnesium ions to the enzyme-GTP complex, allowing us to observe the GTP-enzyme complex trapped in the crystal prior to cleavage. Once the complex is challenged with a high enough concentration of manganese ions, the complex is able to undergo catalysis. Comparison of the molecular surface of the open and closed forms of the enzyme (Figure 5) reveals that after domain closure, most of the GTP-binding site is blocked off from the solvent, with the exception of an opening that could provide access for the incoming cation. Hence, the closing of the protein both ensures that the GTP binds in a conformation that is ready for nucleophilic attack by Lys-82 and creates the binding site for the incoming cation. However, after formation of the GMP adduct, the enzyme must open up to provide access for the incoming mRNA substrate, since this site is blocked off in the closed form of the enzyme.
|
||||
There is some evidence that other members of this nucleotidyltransferase
family, such as DNA ligase, undergo similar conformational changes
during catalysis. The structural similarity between the capping
enzymes and ATP-dependent DNA ligases was predicted from sequence
alignments (![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
It is likely that the structures of cellular RNA guanylyltransferases
will resemble that of the PBCV-1 enzyme. An alignment of the
sequence of the PBCV-1 protein with the sequences of the capping
enzymes encoded by S. cerevisiae (![]()
![]()
![]()
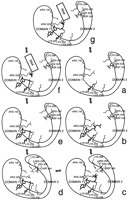
We can now propose a general mechanism for phosphoryl transfer in this family of nucleotidyltransferases (Figure 6 and Figure 7), based upon four different crystal structures. In the first step of the reaction, GTP (or ATP in the case of ligase) binds to the open form of the enzyme. The binding of GTP promotes closure of the domains. This closure is stabilized by interactions between the bound nucleotide and residues on domain 2. The domain closure is followed by phosphoryl transfer from GTP to produce the GMP adduct of the enzyme. The cleavage of GTP breaks the linkage between residues in domain 2 and the nucleotide and thus destabilizes the closed form of the enzyme, which is then able to open up again to release pyrophosphate and expose the RNA-binding site, allowing RNA to bind to the enzyme ready for guanylation.
|
||||
|
||||
The structures of the capping enzyme presented here confirm the similarity between the NTP-binding domains of mRNA-capping enzymes and ATP-dependent DNA ligases, indicating a conservation of function for the conserved motifs in both structures. The difference in conformation observed for the two GTP complexes provides direct evidence for a significant conformational change during nucleotidyl transfer in the capping enzymes, which is likely to be similar in other members of this enzyme family. However, it is evident that this is not the whole story and that there are still many unanswered questions about nucleotidyl transfer from the enzymes to their RNA or DNA substrates. Additional structural information, particularly about a complex of a capping enzyme or a ligase complexed with its nucleic acid substrate, should shed further light on this step in the enzyme mechanism.
| Experimental Procedures |
|---|
| Summary | Introduction | Results and Discussion | Procedures | References |
Protein was prepared and crystallized as described previously (![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() positions is 0.44 Å for domain 1 but 8.5 Å for domain
2. However, when domains 2 alone from each molecule are superimposed,
the rms deviation for C
positions is 0.44 Å for domain 1 but 8.5 Å for domain
2. However, when domains 2 alone from each molecule are superimposed,
the rms deviation for C![]() positions is only 1.4 Å. These data indicate that the
domain movement is largely a consequence of a rigid body rotation
between the two conformations of the protein rather than an
alteration of the secondary structure.
positions is only 1.4 Å. These data indicate that the
domain movement is largely a consequence of a rigid body rotation
between the two conformations of the protein rather than an
alteration of the secondary structure.
The GMP adduct was prepared by harvesting crystals as described
previously (![]()
| Acknowledgments |
|---|
Correspondence regarding this paper should be addressed to D. B. W. We wish to thank Synchrotron Radiation Source (Daresbury) for access to synchrotron radiation facilities. We also thank H. Subramanya for advice on density averaging, S. Islam and M. Sternberg for providing their graphics program PREPPI, K. Ho for preparing the capping enzyme overexpressing cell line, and D. Barford for helpful discussions. This project was funded by the Wellcome Trust and the Medical Research Council.
Received February 14, 1997; revised March 26, 1997.
| References |
|---|
| Summary | Introduction | Results and Discussion | Procedures | References |
Ahola, T., and Kaariainen, L. (1995). Reaction in alphavirus mRNA capping: formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. USA 92, 507-511.[Medline]
Brünger, A.T. (1992). Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472-474.
Brünger, A.T., Karplus, M., and Petsko, G.A. (1989). Crystallographic refinement by simulated annealing: application to crambin. Acta Cryst. A45, 50-61.
Carson, M. (1991). Ribbons 2.0. J. Appl. Cryst. 24, 958-961.
Collaborative Computing Project No. 4 (1994). The CCP4 suite: programs for protein crystallography. Acta Cryst. D50, 760-763.
Cong, P., and Shuman, S. (1993). Covalent catalysis in nucleotidyl transfer. A KTDG motif essential for enzyme-GMP complex formation by mRNA capping enzyme is conserved at the active sites of RNA and DNA ligases. J. Biol. Chem. 268, 7256-7260.[Medline]
Cong, P., and Shuman, S. (1995). Mutational analysis of messenger-RNA capping enzyme identifies amino acids involved in GTP-binding, enzyme-guanylate formation and GMP transfer to DNA. Mol. Cell. Biol. 15, 6222-6231.[Medline]
Doherty, A.J., Ashford, S.R., Subramanya, H.S., and Wigley, D.B. (1996). Characterisation of proteolytic fragments of bacteriophage T7 DNA ligase. Nucl. Acids Res. 24, 2281-2287.
Doherty, A.J., Håkansson, H., Ho, C.K., Shuman, S., and Wigley, D.B. (1997). Crystallisation of the RNA guanyltransferase of Chlorella virus PBCV-1. Acta Cryst. D, in press.,.
Fresco, L.D., and Buratowski, S. (1994). Active site of the mRNA-capping enzyme guanylyltransferase from Saccharomyces cerevisiae: similarity to the nucleotidyl attachment motif of DNA and RNA ligases. Proc. Natl. Acad. Sci. USA 91, 6624-6628.[Medline]
Heaphy, S., Singh, M., and Gait, M.J. (1987). Effect of single amino acid changes in the region of the adenylation site of T4 RNA ligase. Biochemistry 26, 1688-1696.[Medline]
Ho, C.K., van Etten, J.L., and Shuman, S. (1996). Expression and characterization of an RNA capping enzyme encoded by Chlorella virus PBCV-1. J. Virol. 70, 6658-6664.[Medline]
Jones, T.A., Zou, J-Y., Cowan, S.W., and Kjeldgaard, M. (1991). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. A47, 110-119.
Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283-291.
Leatherbarrow, R.J., Fersht, A.R., and Winter, G. (1985). Transition state stabilisation in the mechanism of tyrosyl transfer RNA synthetase revealed by protein engineering. Proc. Natl. Acad. Sci. USA 82, 7840-7844.[Medline]
Li, Y., Lu, Z.Q., Burbank,
P.E., Kutish, G.F., Rock, D.L., and Vanetten, J.L. (1995). Analysis of
43 KB of the Chlorella virus PBCV-1 330KB genome![]() Map
positions 45-88. Virology 212, 134-150.[Medline]
Map
positions 45-88. Virology 212, 134-150.[Medline]
Nicholls, A., and Honig, B.J. (1991). A rapid finite-difference algorithm, utilizing successive over-relaxation to solve the Poisson-Boltzmann equation. J. Comput. Chem. 12, 435-445.
Otwinowski, Z. (1993). Oscillation data reduction program. In Data Collection and Processing, S.E.R.C. Daresbury DL/SC1/R34, pp. 56-62.
Schwer, B., and Shuman, S. (1994). Mutational analysis of yeast mRNA capping enzyme. Proc. Natl. Acad. Sci. USA 91, 4328-4332.[Medline]
Sheldrick, G. (1992). In Crystallographic Computing 5, D. Moras, A.D. Podjarny, and J.C. Thierry, eds. (Oxford, U.K.: Oxford University Press), pp. 145-157.
Shibagaki, Y., Itoh, N., Yamada, H., Nagata, S., and Mizumoto, K. (1992). mRNA capping enzyme: isolation and characterization of the gene encoding mRNA guanylyltransferase subunit from Saccharomyces cerevisiae. J. Biol. Chem. 267, 9521-9528.[Medline]
Shuman, S. (1995). Capping enzyme in eukaryotic mRNA synthesis. Prog. Nucl. Acids Res. Mol. Biol. 50, 101-129.
Shuman, S. (1996). Closing the gap on DNA ligase. Structure 4, 653-656.[Medline]
Shuman, S., and Hurwitz, J. (1981). Mechanism of mRNA capping by vaccinia virus guanylyltransferase: characterization of an enzyme-guanylate intermediate. Proc. Natl. Acad. Sci. USA 78, 187-191.[Medline]
Shuman, S., Liu, Y., and Schwer, B. (1994). Covalent catalysis in nucleotidyl transfer reactions: essential motifs in Saccharomyces cerevisiae RNA capping enzyme are conserved in Schizosaccharomyces pombe and viral capping enzymes and among polynucleotide ligases. Proc. Natl. Acad. Sci. USA 91, 12046-12050.[Medline]
Shuman, S., and Ru, X.-M. (1995). Mutational analysis of vaccinia DNA ligase defines residues essential for covalent catalysis. Virology 211, 73-83.[Medline]
Shuman, S., and Schwer, B. (1995). RNA capping enzyme and DNA ligase: a superfamily of covalent nucleotide transferases. Mol. Microbiol. 17, 405-410.[Medline]
Subramanya, H.S., Doherty, A.J., Ashford, S.R., and Wigley, D.B. (1996). Crystal structure of an ATP-dependent DNA ligase from bacteriophage T7. Cell 85, 607-614.[Summary/Full Text]
Yamada-Okabe, T., Shimmi, O., Doi, R., Mizumoto, K., Arisawa, M., and Yamada-Okabe, H. (1996). Isolation of the mRNA-capping enzyme and ferric-reductase-related genes from Candida albicans. Microbiology 141, 2515-2523.